



1. Innledende forståelse
På dette stadiet må vi forstå noen konsepter og terminologi, for å unngå å gjøre feil foran våre seniorer, for eksempel:
Spørsmål: Hva er forskjellen mellom RT-PCR, qPCR, sanntids-PCR og sanntids-RT-PCR?
Svar: RT-PCR er revers transkripsjons-PCR(revers transkripsjon PCR, RT-PCR), som er en mye brukt variant av polymerasekjedereaksjon (PCR).I RT-PCR blir en RNA-streng reverstranskribert til komplementært DNA, som deretter brukes som en mal for DNA-amplifisering ved PCR.
Sanntids-PCR og qPCR(Quantitative Real-ltime-PCR) er det samme, begge er sanntids kvantitativ PCR, noe som betyr at hver syklus av PCR har sanntidsdataposter, slik at antall startmaler kan justeres nøyaktig analyse.
Selv om både sanntids-PCR (sanntidsfluorescerende kvantitativ PCR) og omvendt transkripsjons-PCR (revers transkripsjons-PCR) ser ut til å bli forkortet til RT-PCR, er den internasjonale konvensjonen: RT-PCR refererer spesifikt til revers transkripsjonPCR , sanntids PCR er generelt forkortet til qPCR (kvantitativ sanntids PCR).
Og sanntids RT-PCR (RT-qPCR), det er omvendt transkripsjons-PCR kombinert med den fluorescerende kvantitative teknologien: skaff først cDNA (RT) fra RNA revers transkripsjon, og bruk deretter sanntids PCR for kvantitativ analyse (qPCR).De fleste laboratorier driver med RT-qPCR, det vil si forskning på RNA-uttrykk nedregulering, så qPCR som alle snakker om i laboratoriet refererer faktisk til RT-qPCR, men glem ikke at det fortsatt er mange DNA-tester i kliniske applikasjoner.Kvantitativ analyse, for eksempel påvisning av hepatitt B-virus HBV.
Spørsmål: Etter å ha lest mye fluorescerende kvantitativ PCR, hvorfor skal det amplifiserte fragmentet kontrolleres innenfor området 80-300bp?
Svar: Lengden på hver gensekvens er forskjellig, noen er flere kb, noen er hundrevis av bp, men vi trenger bare å kreve at produktlengden skal være 80-300bp når vi designer primere, for korte eller for lange er ikke egnet for fluorescerende kvantitativ PCR-deteksjon.Produktfragmentet er for kort til å kunne skilles fra primer-dimeren.Lengden på primer-dimeren er ca. 30-40bp, og det er vanskelig å skille om det er en primer-dimer eller et produkt hvis den er mindre enn 80bp.Hvis produktfragmentet er for langt, over 300 bp, vil det lett føre til lav amplifikasjonseffektivitet og kan ikke effektivt oppdage mengden av genet.
Når du for eksempel teller hvor mange som er i et klasserom, trenger du bare å telle hvor mange munner det er.Det samme gjelder når du oppdager gener, du trenger bare å oppdage en bestemt sekvens av et gen for å representere. Hele sekvensen vil gjøre det.Hvis du vil telle folk, må du telle både munn og nese, ører og briller, og det er lett å gjøre feil.
For å utvide, i biologisk forskning, er det mange forskningstilfeller fra punkt til område, fordi gensekvensen til en hvilken som helst art er veldig lang, det er unødvendig og umulig å måle alle fragmenter, for eksempel bakteriell 16S-sekvensering, som er å utføre den konservative sekvensen av bakterier Analyser for å utlede antallet av en viss populasjon av bakterier.
Spørsmål: Hva er den optimale lengden for qPCR-primerdesign?
Svar: Generelt sett er primerlengden omtrent 20-24bp, noe som er bedre.Selvfølgelig må vi ta hensyn til TM-verdien til primeren når vi designer primeren, fordi dette er relatert til den optimale annealingstemperaturen.Etter mange eksperimenter har det blitt bevist at 60°C er en bedre TM-verdi.Hvis glødingstemperaturen er for lav, vil det lett føre til uspesifikk forsterkning.Hvis annealingstemperaturen er for høy, vil amplifikasjonseffektiviteten være relativt lav, toppen av amplifikasjonskurven vil starte senere, og CT-verdien vil bli forsinket.
Spørsmål: Hvordan er fargemetoden forskjellig fra probemetoden?
Svar: FargemetodeNoen fluorescerende fargestoffer, slik som SYBR Green Ⅰ, PicoGreen, BEBO, etc., avgir ikke lys av seg selv, men vil avgi fluorescens etter binding til det mindre sporet av dobbelttrådet DNA.Derfor, i begynnelsen av PCR-reaksjonen, kan ikke maskinen oppdage det fluorescerende signalet.Når reaksjonen når annealing-forlengelsesstadiet, åpnes dobbeltstrengen, og en ny streng syntetiseres under påvirkning av DNA-polymerase, og det fluorescerende molekylet binder seg til dsDNA-minor-sporet.Etter hvert som antallet PCR-sykluser øker, kombineres flere og flere fargestoffer med dobbelttrådet DNA, og det fluorescerende signalet forbedres også kontinuerlig.Fargemetoden brukes hovedsakelig i vitenskapelig forskning.
PS: Vær forsiktig når du gjør eksperimentet, fargestoffet må kombineres med menneskelig DNA, pass på å gjøre det om til en fluorescerende person.
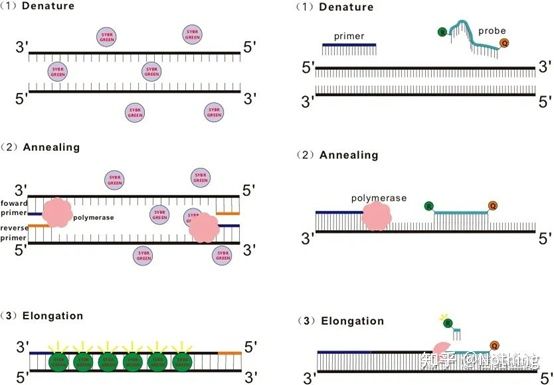
Fargemetode (venstre) Probemetode (høyre)
PS: Vær forsiktig når du gjør eksperimentet, fargestoffet må kombineres med menneskelig DNA, pass på å gjøre det om til en fluorescerende person.
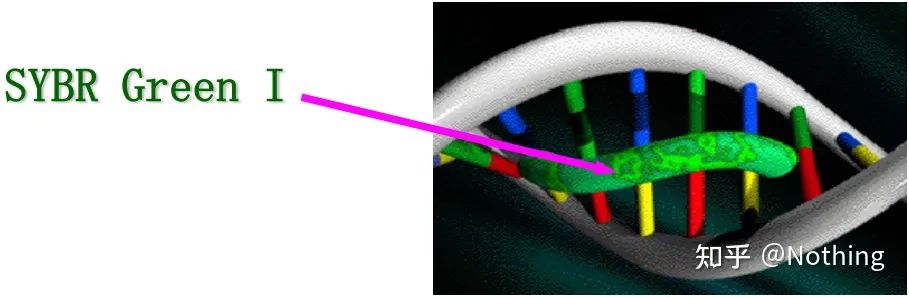
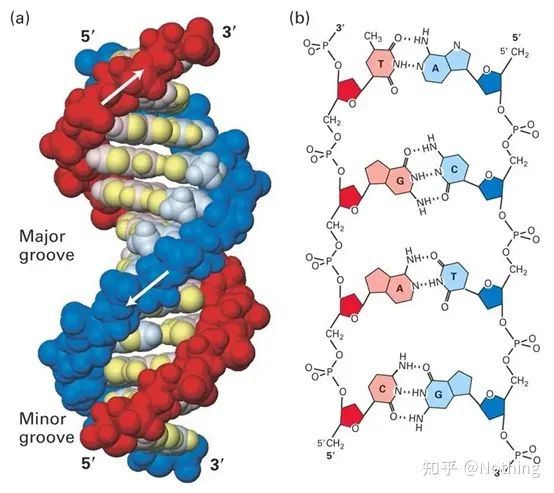
SYBR Grønn Ⅰ binder seg til den mindre rillen i DNA
ProbemetodeTaqman-sonden er den mest brukte hydrolysesonden.Det er en fluorescerende gruppe i 5′-enden av proben, vanligvis FAM, og selve proben er en sekvens komplementær til målgenet.Det er en fluorescerende slukkegruppe i 3′-enden.I henhold til prinsippet om fluorescensresonansenergioverføring (Förster resonance energy transfer, FRET), når reporterfluorescensgruppen (donorfluorescerende molekyl) og den slukkende fluorescerende gruppen (akseptorfluorescerende molekyl) er eksitert når spektrene overlapper og avstanden er svært nær (7-10 fluorescerende fluorescerende molekyl) akseptormolekylet, mens autofluorescensen er svekket.Derfor, i begynnelsen av PCR-reaksjonen, når proben er fri og intakt i systemet, vil ikke reporterfluorescensgruppen avgi fluorescens.Ved annealing binder primeren og proben seg til malen.Under forlengelsesstadiet syntetiserer polymerasen kontinuerlig nye kjeder.DNA-polymerase har 5′-3′ eksonukleaseaktivitet.Når den når proben, vil DNA-polymerasen hydrolysere proben fra malen, skille reporter-fluorescerende gruppen fra quencher-fluorescerende gruppen og frigjøre det fluorescerende signalet.Siden det er et en-til-en forhold mellom proben og malen, er probemetoden overlegen fargemetoden når det gjelder nøyaktigheten og følsomheten til testen.Probemetoden brukes hovedsakelig i diagnostisering.
Spørsmål: Hva er absolutt kvantifisering?Hva er relativ kvantifisering?
Svar: Absolutt kvantifisering refererer til beregningen av det første kopinummeret til prøven som skal testes med qPCR, for eksempel hvor mange HBV-virus som er i 1 ml blod.Resultatet oppnådd ved relativ kvantifisering er endringen i mengden av målgenet i en spesifikk prøve i forhold til en annen referanseprøve, og genuttrykket er opp- eller nedregulert.
Spørsmål: Vil mengden RNA-ekstraksjon, revers transkripsjonseffektivitet og amplifikasjonseffektivitet påvirke de eksperimentelle resultatene?
Spørsmål: Vil prøvelagring, ekstraksjonsreagenser, reverstranskripsjonsreagenser og lystransmitterende forbruksvarer påvirke de eksperimentelle resultatene?
Spørsmål: Hvilken metode kan korrigere eksperimentelle data?
Når det gjelder disse problemene, vil vi beskrive dem i detalj i de avanserte og avanserte delene nedenfor.
2. Avansert kunnskap
Når det gjelder sanntids fluorescerende kvantitativ PCR, må vi erkjenne realiteten at tusenvis av vitenskapelige forskningsartikler publiseres hvert år, blant dem er den fluorescerende kvantitative PCR-teknologien ikke et lite antall.
Hvis det ikke er noen felles standard for å måle det fluorescerende kvantitative PCR-eksperimentet, kan resultatene variere mye.For samme gen av samme art, med samme prosesseringsmetode, vil deteksjonsresultatene også variere mye, og det vil være vanskelig for etternølere å gjenta de samme resultatene.Du Ingen vet hva som er rett og hva som er galt.
Betyr dette at fluorescerende kvantitativ PCR er en jukseteknologi eller en upålitelig teknologi?Nei, det er fordi fluorescerende kvantitativ PCR er mer sensitiv og mer nøyaktig, og litt feil operasjon vil gi helt motsatte resultater.Et lite tap er tusen mil unna.Forfatteren av artikkelen kan gjentatte ganger bli torturert av anmelderne.Samtidig er anmelderne av tidsskriftet også vanskelige å velge blant ulike eksperimentelle resultater.
Alt i alt peker på mangel på konsensus i sanntids PCR-eksperimenter.For dette formål begynte seniorforskere i industrien å formulere standarder,krever at bidragsytere oppgir noen nødvendige eksperimentelle og databehandlingsdetaljer (inkludert nødvendige data) i artikkelen for å oppfylle disse standardene .
Anmeldere kan bedømme kvaliteten på eksperimentet ved å lese disse detaljene;fremtidige lesere kan også bruke dette til å gjenta eksperimentet eller forbedre eksperimentet.Da er de eksperimentelle resultatene oppnådd på denne måten fulle av informasjon, høy kvalitet og brukbare.
MIBBI (minimumsinformasjon for biologiske og biomedisinske undersøkelser -http://www.mibbi.org) ble til.MIBBI er et prosjekt som gir standarder for eksperimenter.Den er publisert i naturen.Dette prosjektet er rettet mot ulike biologiske eksperimenter, inkludert cellebiologi, Microarray, qPCR vi skal diskutere nå, etc., og legger opp til hver type eksperiment ved innsending av manuskripter.Denne informasjonen bør gis til enhver tid.
I MIBBI-prosjektet er det to artikler knyttet til fluorescerende kvantitativ PCR, nemlig:
·RDML (Real-Time PCR Data Markup Language) – en strukturert språk- og rapporteringsveiledning for kvantitative PCR-data i sanntid;
·MIQE (Minimumsinformasjon for publisering av kvantitative sanntids PCR-eksperimenter) – minimumsinformasjon for publisering av artikler om kvantitative PCR-eksperimenter i sanntid.
Først, la oss snakke om RDML, terminologispesifikasjonen.
Hvis det ikke finnes en standarddefinisjon for alt, er det umulig å fortsette diskusjonen, og derfor er begrepsforklaringen så viktig på eksamen.
Terminologien som brukes i det fluorescerende kvantitative PCR-eksperimentet inkluderer følgende innhold.QIAGEN har laget den beste oppsummeringen for oss.Følgende er alle tørrevarer .
Amplifikasjonskurve
Amplifikasjonskurven refererer til kurven laget under PCR-prosessen, med syklusnummeret som abscisse og sanntidsfluorescensintensiteten under reaksjonen som ordinaten.
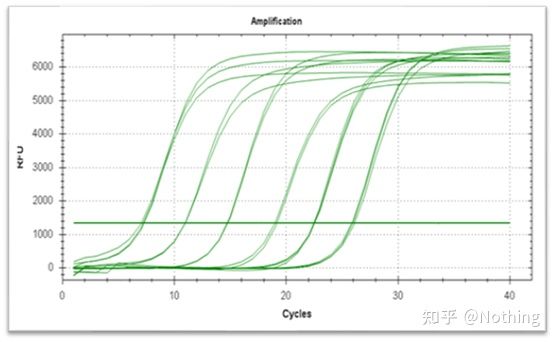
En utmerket amplifikasjonskurve bør ha følgende egenskaper: grunnlinjen er flat eller svakt redusert, og det er ingen åpenbar oppadgående trend;kurvens bøyningspunkt er klart, og helningen til den eksponentielle fasen er proporsjonal med amplifikasjonseffektiviteten.Jo større helning, desto høyere forsterkningseffektivitet;den generelle amplifikasjonskurven. Parallellen er god, noe som indikerer at amplifikasjonseffektiviteten til hvert rør er lik;den eksponentielle fasen av amplifikasjonskurven til prøver med lav konsentrasjon er åpenbar.
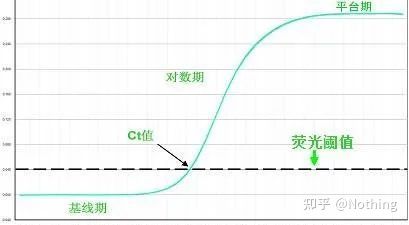
Grunnlinje (grunnlinje)
Grunnlinjen er støynivået for den tidlige syklusen, vanligvis målt mellom 3. og 15. syklus, fordi økningen i fluorescensverdien forårsaket av amplifikasjonsproduktet ikke kan påvises i denne perioden.Antallet sykluser som brukes til å beregne grunnlinjen kan varieres og må kanskje reduseres hvis høye malmengder brukes eller hvis ekspresjonsnivået til målgenet er høyt.
Innstilling av basislinjen krever visning av fluorescensdata fra linearitetsforsterkningskurven.Grunnlinjen er satt slik at veksten av amplifikasjonskurven begynner med et syklustall som er større enn topptallet for baselinesyklusen.Grunnlinjer må settes individuelt for hver målsekvens.De gjennomsnittlige fluorescensverdiene oppdaget i de tidlige syklusene må trekkes fra fluorescensverdiene oppnådd i de amplifiserte produktene.De nyeste versjonene av ulike Real-Time PCR-programvare tillater automatisk optimalisering av grunnlinjeinnstillinger for individuelle prøver.
I løpet av de første par syklusene av PCR-amplifikasjonsreaksjonen endres ikke fluorescenssignalet mye.Å nærme seg en rett linje kalles grunnlinjen, men hvis vi ser nøye på de første syklusene, ser vi at innenfor grunnlinjen er det som skjer på bildet nedenfor.
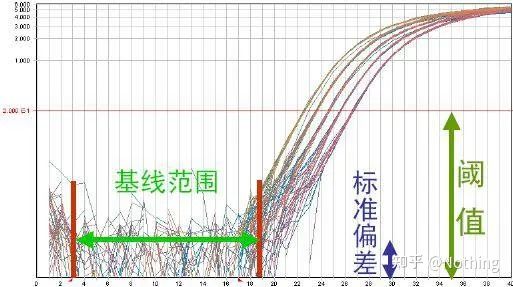
Bakgrunn Bakgrunn refererer til
den uspesifikke fluorescensverdien i reaksjonen.For eksempel: ineffektiv fluorescensslukking;eller et stort antall dobbelttrådete DNA-maler på grunn av bruken av SYBR Green.Bakgrunnskomponentene til signalet fjernes matematisk av Real-Time PCR-programvarealgoritmen.
Reporter signal
Reportersignal refererer til det fluorescerende signalet generert av SYBR Green eller fluorescensmerkede sekvensspesifikke prober under sanntids-PCR.
Normalisert reportersignal (RN)
RN refererer til fluorescensintensiteten til reporterfargestoffet delt på fluorescensintensiteten til det passive referansefargestoffet målt ved hver syklus.
Passiv referansefarge
I noen sanntids-PCR-er,det fluorescerende fargestoffet ROX brukes som en intern referanse for å normalisere det fluorescerende signalet.Den korrigerer for variasjoner på grunn av unøyaktig pipettering, brønnposisjon og fluorescensfluktuasjoner brønn for brønn.

Fluorescensterskelen (terskel)
ble justert over bakgrunnsverdien og betydelig under platåverdien til amplifikasjonskurven.Den må ligge i det lineære området av amplifikasjonskurven, som representerer det log-lineære området for PCR-deteksjon.Terskler bør settes i log-amplifikasjonskurvevisningen slik at den log-lineære fasen av PCR er lett identifiserbar.Hvis det er flere målgener i sanntids-PCR, må terskelen settes for hvert mål.Generelt brukes fluorescenssignalet til de første 15 syklusene av PCR-reaksjonen som fluorescensbakgrunnssignalet, og fluorescensterskelen er 10 ganger standardavviket til fluorescenssignalet for de første 3 til 15 PCR-syklusene, og fluorescensterskelen settes i PCR-amplifiseringsfasen.Generelt har hvert instrument sin fluorescensterskel satt før bruk.
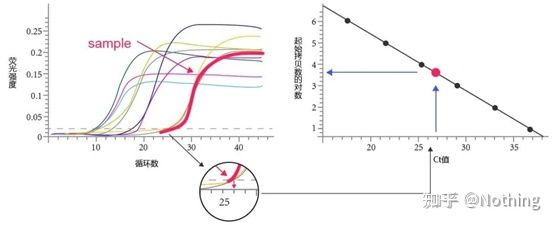
Syklusterskel (CT) eller Krysningspunkt (CP)
Syklusen der amplifikasjonskurven krysser terskelen (dvs. punktet der fluorescensdeteksjonen øker betydelig).CT kan være en brøkdel og mengden startmal kan beregnes.CT-verdien representerer antall sykluser som oppleves når det fluorescerende signalet i hvert PCR-reaksjonsrør når den innstilte terskelen.Det er et lineært forhold mellom CT-verdien til hver mal og logaritmen til det første kopinummeret til malen,høyere det opprinnelige kopinummeret, jo mindre er CT-verdien, og omvendt.En standardkurve kan lages ved å bruke en standard med kjent startkopinummer, hvor abscissen representerer CT-verdien, og ordinaten representerer logaritmen til startkopitallet.Så lenge CT-verdien til den ukjente prøven oppnås, kan derfor det første kopinummeret til prøven beregnes fra standardkurven.
ΔCT-verdi
ΔCT verdi beskriverforskjellen mellom målgenet og den tilsvarende endogene referansegenets CT-verdi, for eksempel et husholdningsgen, og brukes til å normalisere mengden mal som brukes:
⇒ΔCT = CT (målgen) – CT (endogent referansegen)
ΔΔCT-verdi
ΔΔCT-verdien beskriver forskjellen mellom den gjennomsnittlige ΔΔCT-verdien til en prøve av interesse (f.eks. stimulerte celler) og den gjennomsnittlige ΔΔCT-verdien til en referanseprøve (f.eks. ustimulerte celler).Referanseprøven kalles også kalibreringsprøven, og alle andre prøver er normalisert til denne for relativ kvantifisering:
⇒ΔΔCT = gjennomsnittlig ΔCT (utvalg av interesse) – gjennomsnittlig ΔCT (referanseutvalg)
Endogene referansegener (endogene referansegener)
Ekspresjonsnivåene til endogene referansegener, slik som husholdningsgener (husholdningsgener), skiller seg ikke mellom prøvene.Sammenligning av CT-verdiene til referansegenet med målgenet gjør at ekspresjonsnivået til målgenet kan normaliseres til mengden input-RNA eller cDNA (se avsnittet om ΔCT-verdier ovenfor).
Interne referansegener korrigerer formulig RNA-nedbrytning eller tilstedeværelse av enzymhemmere i RNA-prøver, samt variasjoner i RNA-innhold, revers transkripsjonseffektivitet, nukleinsyregjenvinning og prøvehåndtering.For å velge det eller de optimale referansegenene, modifiserte vi algoritmen for å tillate valg av den optimale referansen avhengig av den eksperimentelle innstillingen.
Indre kontroll
En kontrollsekvens som blir amplifisert i samme reaksjon som målsekvensen og probet med en annen probe (dvs. utfører dupleks PCR).Internkontroll brukes ofte for å utelukke mislykkede amplifikasjoner, for eksempel når målsekvensen ikke oppdages.
Kalibreringsprøve
En referanseprøve (for eksempel renset RNA fra en cellelinje eller vev) brukt i relativ kvantifisering for å sammenligne alle andre prøver for å bestemme det relative ekspresjonsnivået til et gen.Kalibreringsprøven kan være en hvilken som helst prøve, men er vanligvis en kontroll (for eksempel en ubehandlet prøve eller en prøve fra tidspunkt null i eksperimentet).
Positive kontroller
bruke kontrollreaksjoner medkjente mengder mal.Positive kontroller brukes ofte for å kontrollere at et primersett eller primer-probesett fungerer som det skal, og at reaksjonen er satt opp riktig.
Ingen malkontroll (NTC)
En kontrollreaksjon som inneholder alle de nødvendige komponentene i amplifikasjonsreaksjonen bortsett fra malen, som vanligvis erstattes med vann.Bruken av NTC kan finne kontamineringen forårsaket av reagenskontaminering eller fremmed DNA, og dermed sikre autentisiteten og påliteligheten til deteksjonsdataene.Amplifisering av NTC-kontrollen indikerer forurensning.
Ingen RT-kontroll (NRT)
RNA-ekstraksjonsprosessen kan inneholde gjenværende genomisk DNA, som er ekstremt skadelig og er den skyldige som påvirker datakvaliteten og den naturlige fienden til qPCR, så når du designer eksperimenter, må den utformes for kun å forsterke RNA-deteksjon.Det er to måter, den ene er å designe primere på tvers av introner, den andre er å fjerne DNA fullstendig, hvilken er bedre, som vil bli diskutert senere.NTR-kontrollen er et magisk speil for å oppdage DNA-forurensning.Hvis det er forsterkning, betyr det at det er forurensning.
Standarder
Standarder er prøver med kjent konsentrasjon eller kopiantall som brukes til å konstruere en standardkurve.For å sikre stabiliteten til standarden blir genfragmentet vanligvis klonet inn i plasmidet og brukt som standard.
Standardkurven
er vanligvis fortynnet i minst 5 konsentrasjonsgradienter med standardproduktet i henhold til doblingsforholdet, og 5 punkter tegnes i koordinatene til CT-verdi og kopitall, og punktene kobles sammen for å danne en linje for å generere en standardkurve.For hver standardkurve må dens gyldighet kontrolleres.Helningsverdien faller mellom –3,3 og –3,8, og hver konsentrasjon utføres i triplikat.Punkter som er vesentlig forskjellig fra andre punkter bør forkastes.CT-verdien til prøven som skal testes bringes inn i standardkurven, og uttrykksnivået til prøven som skal testes kan beregnes.
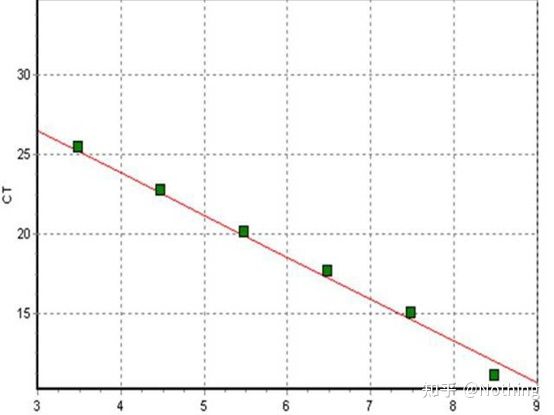
CT-verdien til prøven som skal testes bringes inn i standardkurven, og det første kopinummeret til prøven som skal testes kan beregnes.
Effektivitet og helning
Helningen til standardkurven representerer effektiviteten til sanntids PCR.
·En helning på -3,322 indikerer at PCR-amplifikasjonseffektiviteten er 1, eller 100 % effektiv, og mengden PCR-produkt dobles ved hver syklus.
·En helning mindre enn –3,322 (f.eks. –3,8) indikerer en PCR-effektivitet
·En helning større enn –3,322 (f.eks. –3,0) indikerer at PCR-effektiviteten ser ut til å være større enn 100 %, noe som er nysgjerrig, hvordan kan en PCR-syklus generere mer enn det dobbelte av det amplifiserte produktet?Denne situasjonen oppstår i den ikke-lineære fasen av PCR-reaksjonen, det vil si at det er en stor mengde ikke-spesifikk amplifikasjon.
smeltekurve
Etter at qPCR-amplifikasjonen er fullført, varmes PCR-produktet opp.Når temperaturen stiger, smelter det dobbelttrådete amplifikasjonsproduktet gradvis, noe som resulterer i en reduksjon i fluorescensintensitet.Når en viss temperatur (Tm) er nådd, vil et stort antall produkter smelte.Fluorescensen synker kraftig.Ulike PCR-produkter har forskjellige Tm-verdier og forskjellige smeltetemperaturer, slik at spesifisiteten til PCR kan identifiseres.
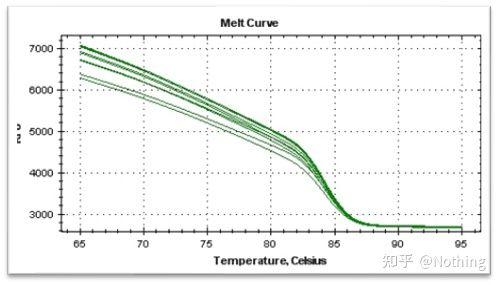
Smeltekurve (derivert kurve)
Smeltekurven er utledet for å danne et toppkart, som mer intuitivt kan vise situasjonen til PCR-produktfragmenter.Siden smeltetemperaturen er Tm-verdien til DNA-fragmentet, kan noen parametere som påvirker Tm-verdien til DNA-fragmentet bedømmes, slik som fragmentstørrelse, GC-innhold osv. Generelt sett, i henhold til våre primerdesignprinsipper,lengden på det forsterkede produktet er i området 80-300 bp, så smeltetemperaturen bør være mellom 80°C og 90°C.
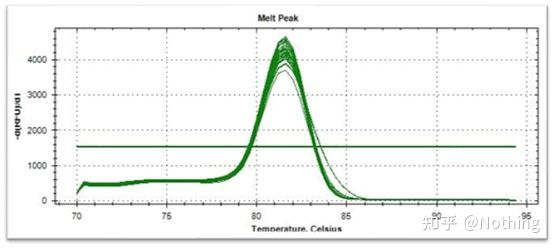
Tolkning av smeltekurven: Hvis den eneste hovedtoppen vises mellom 80°C-90°C, betyr det at den fluorescerende kvantitative PCR er perfekt;hvis hovedtoppen vises mellom 80°C-90°C og diverse topper vises under 80°C, vurderes primerdimeren i utgangspunktet.Du kan prøve å øke utglødningstemperaturen for å løse det;hvis hovedtoppen vises mellom 80°C-90°C, og den øvrige toppen vises igjen når temperaturen stiger, anses det i utgangspunktet at det er DNA-kontaminering, og DNA må fjernes i den innledende fasen av eksperimentet.
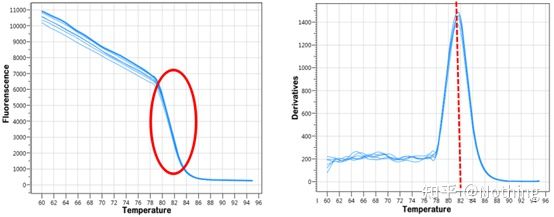
Selvfølgelig er det fortsatt noen unormale situasjoner, som vil bli brutt ned en etter en nedenfor.
3. Avansert kunnskap
For å gjøre qPCR, må jeg si MIQE,Minimum informasjonfor publisering avKvantitativSanntids PCREksperimenter – minimumsinformasjonen for å publisere artikler om sanntids kvantitativ PCReksperimenter.For å forenkle alles forståelse, vil vi forenkle nøkkelinnholdet.
Du kan søke i originalteksten til MIQE på Internett, og det viktigste er at den fastsetterdatasjekkliste som må oppgis ved publisering av en artikkel .
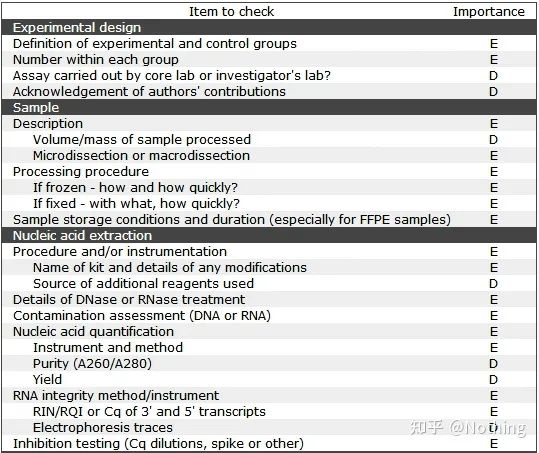
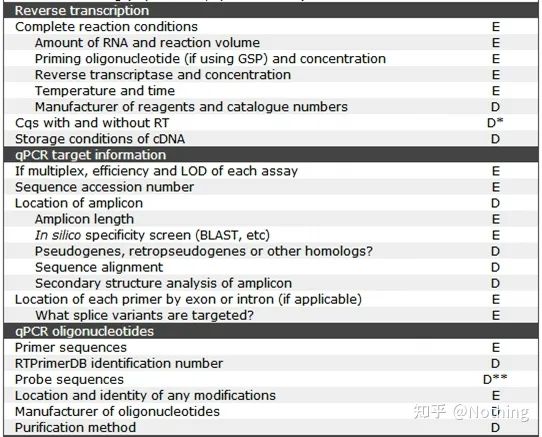
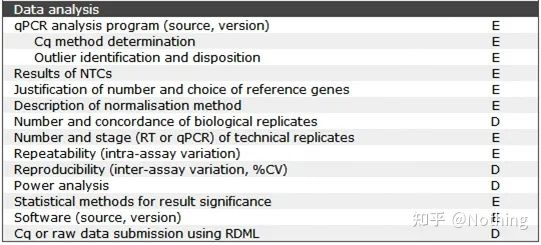
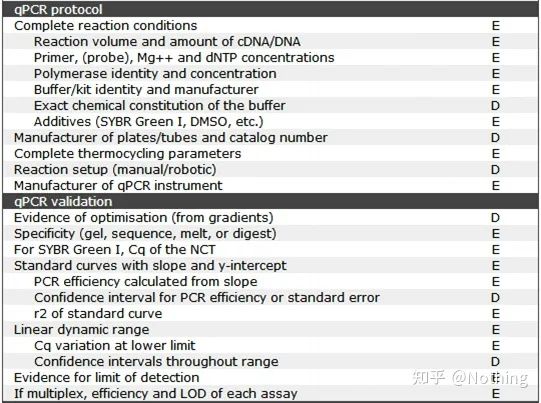
Anmeldere kan bedømme kvaliteten på eksperimentet ved å lese disse detaljene;fremtidige lesere kan også bruke dette til å gjenta eller forbedre eksperimentet.
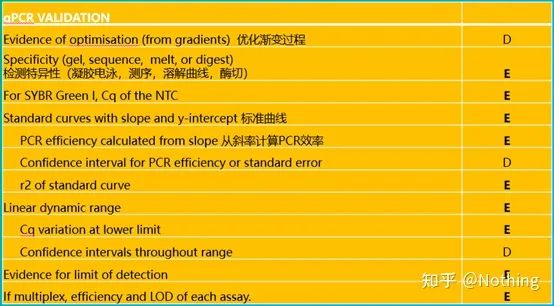
Det er verdt å merke seg at i denne listen er viktigheten av hver liste merket med henholdsvis E eller D.Hva betyr det?E: viktig informasjon (må sendes inn);D: ønskelig informasjon (gi så mye som mulig).
MIQE (1) – Eksperimentell design
Mange skurker som har fullført forsvaret etter å ha fullført hovedstudiene, vil ikke vite hvordan de skal designe et eksperiment uavhengig, åpne notatbøkene og gjøre det læreren ber dem om.Som et resultat var det eksperimentelle designet ikke strengt, og redaksjonen til magasinet sa at de ønsket å lage dette bildet og det bildet, så de gjorde det i en døs.Slik lages skurkene!

Nærmere hjemmet er det første prinsippet i eksperimentet å bestemmestrengheten til den eksperimentelle logikken.Det mest grunnleggende er det eksperimentelle designet, og det viktigste med det eksperimentelle designet er hvordan man setter målprøven, referanseprøven (kontroll) og antall repetisjoner, slik at de eksperimentelle dataene kan refereres, sammenlignbare og overbevisende.
Målprøvenrefererer til prøven som krever at vi oppdager målgenet etter en viss behandling.Referanseprøvener prøven uten noen behandling, som ofte omtales som villtype i biologi.
Eksperimentelle replikaterer veldig viktige.Generelt må antallet overbevisende replikater være mer enn tre.Det er nødvendig å skille hva som er biologisk replikasjon og hva som er teknisk replikasjon.
Biologiske replikater: Det samme verifikasjonsforsøket utført med forskjellige materialer (tid, planter, partier, reaksjonsplater).

Biologisk duplisering
La oss ta pesticidbehandling av pepper som et eksempel.Vi ønsker å sprøyte plantevernmidler på de tre plantene til ABC, så er de tre plantene til ABC tre biologiske replikater, og de er det samme verifikasjonsforsøket utført med forskjellige materialer.Men som et eksperiment er det definitivt nødvendig med en kontroll, så vi kan sprøyte en av grenene til plante A for å danne en eksperimentell gruppe av plante A, og ikke sprøyte de andre grenene til plante A for å danne en kontrollgruppe.Gjør det samme for B og C.
Tekniske replikater (tekniske replikater): Det er et gjentatt eksperiment designet for å unngå feil forårsaket av operasjon, som faktisk er et duplikathull inkludert i det samme materialet.Både behandlinger og kontroller må ha replikatinnstillinger (minimum tre) av målgenet og det interne referansegenet.
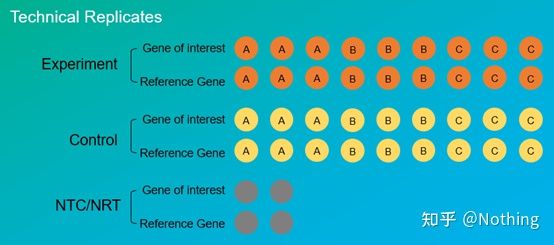
Teknisk repetisjon
Ta paprikaen behandlet med sprøytemidler som et eksempel igjen.For den eksperimentelle gruppen av plante A lagde vi tre PCR-hull på 1, 2 og 3 for henholdsvis målgenet og det interne referansegenet, for å ta gjennomsnittet etter deteksjonen.For kontroll av anlegg A Grupper behandles også på samme måte.Gjør samme behandling for B- og C-planter.Dette er teknisk repetisjon.
Det er verdt å merke seg detdet som kommer inn i statistikken er den biologiske repetisjonen, og den tekniske repetisjonen er å teste om det er noen tilfeldige fenomener i den eksperimentelle prosessen, for å gjøre forsøksresultatene troverdige, det vil si å unngå feil ved å ta gjennomsnittet som vi ofte sier.
Negative kontroller – NTC og NRT
NTC (No-Template Control), en kontroll uten mal, brukes for å verifisere om det eksperimentelle materialet er kontaminert.Vanligvis brukes vann som mal.Hvis det er en fluorescerende reaksjon, indikerer det at nukleinsyreforurensning har skjedd i laboratoriet.
Disse forurensningene kommer fra: urent vann, ukvalifiserte reagenser som inneholder endogent DNA, primerforurensning, laboratorieutstyrsforurensning, aerosolforurensning, etc., må bruke RNase-oppfangere og RNase-hemmere.Aerosolforurensning er det vanskeligste å finne.Tenk deg at laboratoriet ditt er som smog, med ulike nukleinsyrer suspendert i luften.

NRT (No-Reverse Transcriptase), kontrollen uten revers transkripsjon, er det ikke-revers transkriberte RNA som en negativ kontroll, som er kontrollen av gDNA-resten.
Når man gjør genuttrykk, detekteres mengden RNA ved å detektere mengden cDNA etter revers transkripsjon.Hvis det er gDNA-rester når RNA renses, vil det forårsake feil i de eksperimentelle resultatene, fordi de faktiske resultatene som oppnås er gDNA og cDNA.På aggregert nivå, ikke bare cDNA, må gDNA fjernes fullstendig under RNA-ekstraksjon.
MIQE (2) – eksempelinformasjon
Den såkalte prøveinformasjonen betyr at når vi publiserer en artikkel om qPCR, må vi forklare prøveinformasjonen tydelig, noe som er en uunnværlig del av artikkelen.På samme måte, når vi behandler prøver, må vi også regulere vår egen drift for å sikre validiteten til prøvene.

Beskrivelsen av prøven er kun et resultat, og vi bør være mer oppmerksom på materialene som er tatt under hele eksperimentet.
Valg av eksperimentelle materialer
Blodprøver – velg friskt blod, ikke mer enn 4 timer.Celleprøver – velg å samle friske celler i en periode med kraftig vekst.Dyrevev—Velg friskt, kraftig voksende vev.Plantevev – Velg friskt, ungt vev.
Du har sikkert lagt merke til at det er et nøkkelord i disse få setningene: fresh .
For de ovennevnte prøvene er det beste, kostnadseffektive og stabile settet på markedet Foregenes kit, som raskt og enkelt kan trekke ut deres DNA og RNA.
Animal Total RNA Isolation Kit
Plant Total RNA Isolation Kit Plus
Lagring av eksperimentelle materialer
Generelt anbefaler vi ikke oppbevaring av prøver dersom forholdene tillater det.Det er imidlertid mange venner som ikke kan utføre forsøk umiddelbart etter prøvetaking, og noen trenger til og med å frakte tanker med flytende nitrogen til feltet for prøvetaking.
For denne typen hardtarbeidende venn kan jeg bare si at du ikke forstår reagens forbruksvarer.Nå produserer mange reagensforbruksfirmaer reagenser som kan lagre RNA-prøver ved romtemperatur, og du kan velge å bruke dem.Den konvensjonelle lagringsmetoden er lagring av flytende nitrogen, ved bruk av en liten tank for flytende nitrogen som er lett å bære.Etter å ha brakt prøven tilbake til laboratoriet, oppbevar den i et -80°C kjøleskap.

For eksperimenter som involverer RNA, må seksordsprinsippet følges:lav temperatur, ingen enzymer,ogfort .
Konseptet med lav temperatur er lett å forstå;uten enzymer er RNase overalt i verden vi lever i (ellers ville du blitt drept av HIV), så hvordan unngå RNase når du gjør eksperimenter er et veldig viktig konsept;fort,Det er ingen Kung Fu i verden som ikke kan brytes, bare hastighet kan ikke brytes.
Derfor, på en måte, jo kortere ekstraksjonstiden er, jo bedre settet.Hvorfor gjørForegene's kit legger vekt på hastighet, fordi de vet det godt.
PS: Noen jenter gjør eksperimenter veldig nøye, men de er ikke så gode som en slam dunk etter flere års arbeid.De føler at Gud er urettferdig, klager på andre og ser etter livet.Hun forsto det faktisk ikke.Han beskyttet ikke RNA godt, og slam dunk-spilleren var kvikk.Da han gjorde eksperimentet, trodde han at han skulle fullføre slam dunk med tre ganger, fem ganger og to deler, men han gjorde forsøket bra.
Merk: Langsommere, større sjanse for RNase-invasjon.Hvordan trene deg selv til å være rask?Det er ingen måte, bare øv mer.
For ulike eksperimenter og ulike prøver er det fortsatt nødvendig å lese mer litteratur og velge en passende metode for bearbeiding.For prøveinnsamlings- og lagringsprosessen krever MIQE at det må være tydelig skrevet i papiret, slik at anmelderne kan vurdere påliteligheten til papiret, og det er også praktisk for de lamslåtte ungdommene å gjenta eksperimentet ditt.
Selv om biologiske eksperimenter er vanskelige, er de avanserte.Hvis du ikke er forsiktig, kan du velte verden.For eksempel å gjøre SARS til en biokjemisk krise, eller lage hybridris for å redde 1,3 milliarder mennesker.Bildet nedenfor er et kjemisk eksperiment, du bør forstå hvor stolt du er av forskningen din bare ved å se på hans pikklignende utseende.Glem det, ikke svart ham.

MIQE (3) – nukleinsyreekstraksjon.
Nukleinsyreekstraksjon er en stor begivenhet, og alle molekylærbiologiske eksperimenter starter med nukleinsyreekstraksjon.Først av alt, la oss kopiere MIQEs innhold om nukleinsyreekstraksjon.
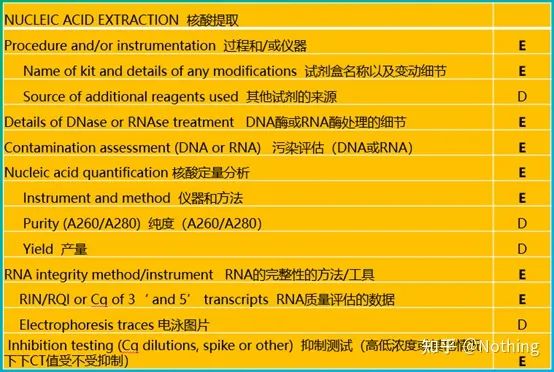
Når du ser på dette skjemaet, kan du ikke holde deg på overflaten.Formen er et dogme.For å være en toppstudent må du spørre hvorfor.Det vesentlige innholdet i denne tabellen er: Fortsettrenheten, integriteten, konsistensen og ekstraksjonsmengden av RNA .
Den første delen avprosess eller instrument er nukleinsyreekstraksjonstrinnet.Hvis du bruker en automatisk nukleinsyreekstraktor for å trekke ut (avansert, vennligst kontakt meg for kjøp), må du angi modellnavnet til instrumentet.

Navnet på settet og
hvilket sett som ble brukt for endringsdetaljene, hvilke spesielle reagenser som ble tilsatt eller hvilke spesielle operasjoner som ble utført, bør forklares tydelig slik at andre enkelt kan gjenta eksperimentet ditt.
Noen legger til noen spesielle reagenser når de trekker ut spesielle prøver, og tenker at dette er deres hemmelige våpen, og forteller ikke det til andre.Mens de holder det hemmelig, mister de også muligheten til å få artikkelen din til å skinne.Ikke vær smart, du må være mer ærlig enn den landet gamle Zhang i vitenskapelig forskning, hvis du vil være smart, vil artikkelen gjøre deg dum.
må huske produktnummeret til settetnår du bestiller settet og skriver artikkelen.Det er vanligvis to numre på settet: Katt – katalognummer (produktnummer, artikkelnummer), Lot – produktpartinummer (Brukes for å indikere hvilken batch produktet kom fra).
I tillegg brukes ofte CAS-nummeret ved bestilling av biokjemiske reagenser, og jeg vil popularisere det sammen.CAS-nummeret er nummeret som gis av American Chemical Society til hvert nytt kjemisk medikament.Vanligvis er tre tall forbundet med en bindestrek.Rushuis CAS-nummer: 7732-18-5.Kjemikalier har ofte flere aliaser, men CAS-nummeret er unikt.Ved bestilling av medisin kan du sjekke CAS-nummeret først.
Nærmere hjemmet, hvorfor må vi beskrive disse tingene tydelig?Faktisk er det også for å sjekke kvaliteten på RNA-ekstraksjon.Bruken av instrumenter og sett vil gjøre RNA-ekstraksjon mer konsekvent.Ekstraksjonsskalaen til vanlige laboratorier er ikke stor, og den kan fås med sett.
Detaljene om DNase- eller RNase-behandling
Det viktige problemet med fluorescerende kvantitativ PCR er å forhindre DNA-kontaminering, og ikke eksperimentere hvis det er forurensning.Derfor er det viktig å oppgi prosessen du brukte for å behandle DNA, for å demonstrere at DNA i den eksperimentelle prosessen er fullstendig og fullstendig fjernet.representert ved et skjematisk diagram.
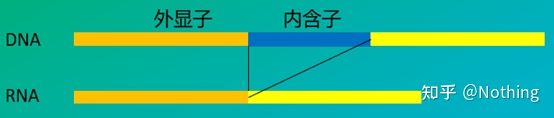
Skjematisk diagram av RNA og DNA
Generelt er metoden for å fjerne DNA å behandle RNA med DNase etter ekstraksjon.Dette er imidlertid relativt gamle metoder.Kommersielle RNA-ekstraksjonssett har vært i stand til å fjerne DNA under ekstraksjonsprosessen uten å tilsette DNase.For eksempel en serie sett fra Foregene .
Merk: Fjerning av DNA under RNA-ekstraksjon er et svært farlig tveegget sverd, som vil forlenge operasjonstiden for RNA-ekstraksjon og øke risikoen for RNA-nedbrytning.I utgangspunktet er det en avveining mellom RNA-utbytte og renhet.
I tillegg er mengden DNase som tilsettes den silikabaserte adsorpsjonskolonnen svært liten, og DNase av høy kvalitet må brukes for å oppnå effekten.Uoptimalisert DNase kan ikke fordøyes raskt og fullstendig.Dette er en test av selgerens tekniske nivå.Selvfølgelig er det enda flere rare kjøpmenn som skryter av at DNA kan fjernes uten DNase.Det kan sies at alle som skryter av at DNA kan fjernes helt uten DNase er en hooligan.DNA er en relativt stabil dobbelttrådet struktur, og den kan ikke utslettes bare ved å snakke og le.
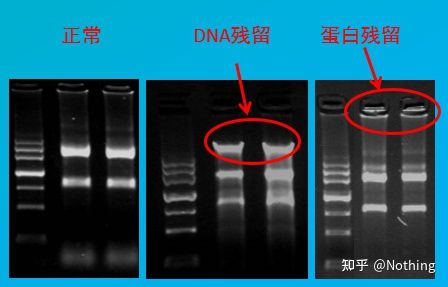
Forurensningsvurdering
vurderingsmetode: elektroforesedeteksjon, 1% agarose, 6V/cm, 15min, lasting 1-3 ul
Kvantitativ nukleinsyreanalyse
måles vanligvis ved hjelp av et UV-spektrofotometer.La meg først popularisere betydningen av de tre verdiene til OD260, OD280 og OD230.
·OD260nm: Det er absorpsjonsbølgelengden til den høyeste absorpsjonstoppen for nukleinsyre, og den best målte verdien varierer fra 0,1 til 1,0.Hvis ikke, fortynn eller konsentrer prøven for å bringe den innenfor rekkevidde.
·OD280nm: Det er absorpsjonsbølgelengden til den høyeste absorpsjonstoppen for protein og fenoliske stoffer.
·OD230nm: Det er absorpsjonsbølgelengden til den høyeste absorpsjonstoppen for karbohydrater.
Deretter skal vi snakke om rollen til hver indikator.For A260 kan den brukes til å måle utbyttet av nukleinsyre.Når OD260=1, dsDNA=50μg/ml, ssDNA=37μg/ml, RNA=40μg/ml.
For renhet må vi se på forholdene som vi vanligvis ser: OD260/280 og OD260/230.
·Rent DNA: OD260/280 er omtrent lik 1,8.Når det er større enn 1,9, indikerer det at det er RNA-forurensning, og når det er mindre enn 1,6, indikerer det at det er protein- og fenolforurensning.
·Rent RNA: 1,7
·OD260/230: Enten det er DNA eller RNA, er referanseverdien 2,5.Når det er mindre enn 2,0, indikerer det at det er forurensning av sukker, salt og organisk materiale.
RNA integritet
Det er veldig viktig å måle integriteten til RNA.Generelt er det nødvendig å gjøre et RNA-denatureringsgeleksperiment for å sjekke om lysstyrken mellom 28S og 18S RNA er et todelt forhold.Når det tredje båndet 5S vises, betyr det at RNA har begynt å brytes ned, bortsett fra virvelløse dyr.

Data for RNA-kvalitetsvurdering: I tillegg til de ovennevnte testene finnes det også noen mer avanserte instrumenttester når det gjelder RNA-integritet, som RQI-integritetstesten til Experion automatiske elektroforesesystem, som kan oppdage om RNA brytes ned usynlig.
I vitenskapelig forskning er fluorescerende kvantitativ PCR en sammenligning mellom målgenet og det interne referansegenet.Derfor, i prosessen med RNA-prøvekonservering, RNA-ekstraksjon, etc., er det primære målet å sikre integriteten til RNA.
Hvordan integriteten til RNA påvirker balansen mellom målgenet og det interne referansegenet kan lett forstås fra figuren nedenfor.Nedbrytning vil føre til genet ufullstendighet, enten det er ufullstendigheten til det interne referansegenet eller ufullstendigheten til målgenet, vil det ha stor innvirkning på dataene.

Skjematisk diagram av målgen og referansegen, må ikke være sant
Inhiberingstest (om CT-verdien er undertrykt under høy eller lav konsentrasjon eller andre forhold)
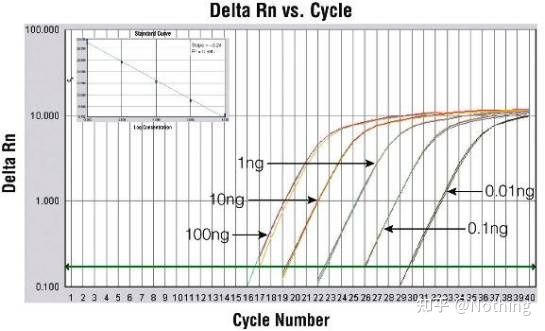
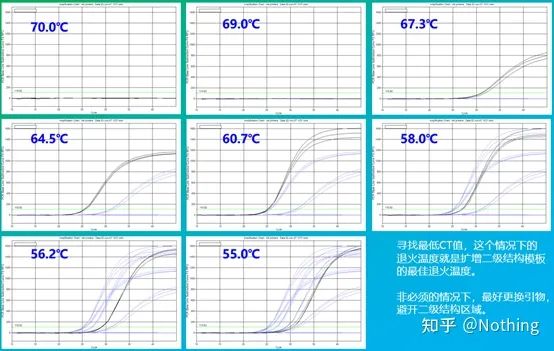
Med denne figuren som et eksempel, er Ct-verdiene til de fem kurvene som følger.Fordelingen av CT-verdier mellom kurvene er ujevn, og Ct-verdiene er forsinket ved høye og lave konsentrasjoner, som er tilfellet med PCR-hemming.
Hovedpoeng: I prosessen med RNA-ekstraksjon må vi forlate misoppfatninger og etablere riktige.
Den gale ideen er: RNA-ekstraksjon forfølger bare utbyttet, og tenker at jo større mengde RNA oppnådd, jo bedre.Faktisk, når vi gjør kvantifisering, hvis antallet gener ikke er veldig stort, trenger vi ikke mye RNA.Mengden RNA du trekker ut er mer enn nok.
Det riktige konseptet er:RNA-ekstraksjon bør forfølge renhet, integritet og konsistens.Renhet kan sikre at den påfølgende revers transkripsjonen ikke hemmes og dataene vil ikke bli påvirket av DNA.Integritet sikrer balansen mellom målsekvenser og interne referanser.Konsistens sikrer stabil prøvebelastning.
MIQE (4) – omvendt transkripsjon
Misforståelse: jakten på høyere prøvevolum.
Riktig konsept: Forsøk konsistens (stabilitet), uavhengig av mengden RNA som er lastet, forblir effektiviteten av revers transkripsjon konsistent, noe som sikrer at forskjeller i cDNA virkelig kan reflektere forskjeller i mRNA.
Vi forklarer denne prosessen med et skjematisk diagram:
Skjematisk diagram av omvendt transkripsjonseffektivitet, er ikke sant
Først av alt må vi forstå forskjellen mellom revers transkripsjonsprosessen og PCR-prosessen.PCR gjennomgår flere oppvarmings- og annealingsprosesser, og målfragmentet vokser eksponentielt;mens revers transkripsjon ikke har denne prosessen, kan vi forestille oss at revers transkripsjon faktisk er en-til-en Under replikasjonsprosessen, så mange deler av RNA
som det er kan få så mange deler av cDNA-informasjon, bør det være forstått nå, fordi fragmenter store og små har blitt omvendt transkribert, og det er umulig å fokusere på ett fragment.Og fordi mengden RNA er relativt liten, er også mengden cDNA som oppnås relativt liten, i motsetning til PCR, som har en amplifikasjonseffekt, så det er i utgangspunktet umulig å oppdage.
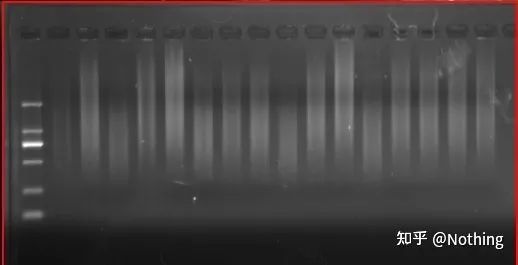
cDNA-elektroforeseresultater
For det andre, ideelt sett utføres revers transkripsjon en-til-en, men ingen revers transkriptase fra noe selskap kan oppnå denne effekten.I utgangspunktet vandrer effektiviteten til de fleste omvendte transkriptaser mellom 30-50%.Hvis dette er tilfelle, vil vi heller ha en relativt stabil revers transkripsjonseffektivitet, som er det vi ønsker å se på figuren: 3 RNA-er får 2 cDNA-er, 6 RNA-er får 4 cDNA-er, så uansett hvor mye prøve som er lastet, er revers-transkripsjonseffektiviteten relativt stabil.Vi ønsker ikke å se situasjonen der revers transkripsjonseffektiviteten er ustabil og høy konsentrasjon hemmes.
Så, hvordan verifisere om den omvendte transkripsjonseffektiviteten er stabil?Metoden er veldig enkel, du trenger bare å gjøre en sammenligningstest: den ene er å reversere transkribere til cDNA etter dobbelfortynning av RNA, og den andre er å gjøre doblefortynning etter reverstranskribering til cDNA, og deretter gjøre qPCR for å se den oppnådde helningen Er den konsistent.Som toppstudent bør du forstå det på sekunder.Som vist under:
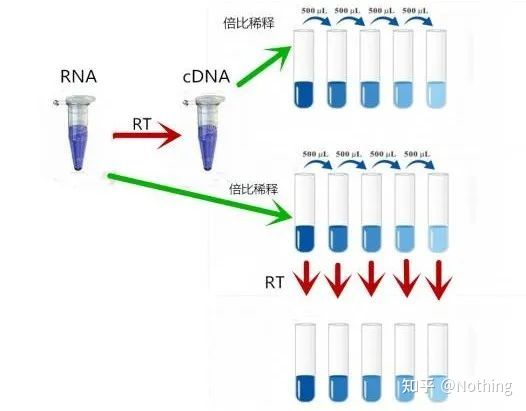
Fortynning av RNA og cDNA for å teste om effektiviteten av revers transkripsjon er stabil
Revers transkriptase og kit
Hvordan kan perfekt fluorescerende kvantitativ PCR ha utmerket revers transkriptase og kit.Revers transkriptase er grovt delt inn i to typer etter kilden, AMV ellerM-MLV, og ytelsen deres er den samme som vist i tabellen.
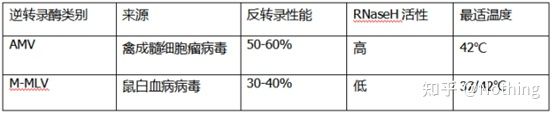
RNase H-aktivitet
RNase H er ribonuklease H, det kinesiske navnet er ribonuklease H, som er en endoribonuklease som spesifikt kan hydrolysere RNA i DNA-RNA-hybridkjeden.RNase H kan ikke hydrolysere fosfodiesterbindingene i enkelt- eller dobbelttrådet DNA eller RNA, det vil si at det ikke kan fordøye enkelt- eller dobbelttrådet DNA eller RNA.Vanligvis brukt i syntesen av den andre strengen av cDNA.
Det er en merkelig ting.Vi sier at revers transkriptase har RNase H-aktivitet, ikke at revers transkriptase inneholder RNase H, og det er kanskje ikke mulig å skille RNase H fra revers transkriptase, kanskje på grunn av konformasjonen til visse grupper i revers transkriptase. Denne aktiviteten er forårsaket av revers transkriptase.
Derfor, uavhengig av den høyere revers transkripsjonseffektiviteten til AMV, reduserer dens RNase H-aktivitet utbyttet av cDNA.Selvfølgelig optimaliserer reagensprodusenter hele tiden produktene sine for å eliminere RNase H-aktivitet i revers transkriptase så mye som mulig for å øke utbyttet av cDNA.
Glødetemperatur
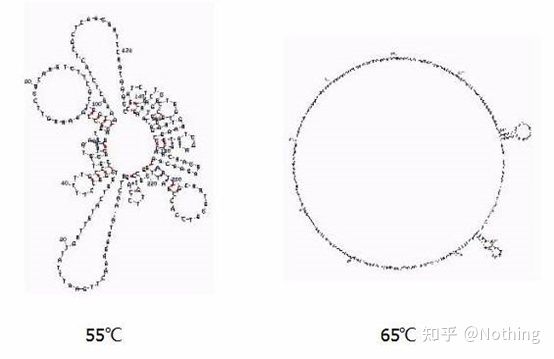
Sekundær struktur av RNA ved forskjellige temperaturer
Se figuren ovenfor for den sekundære strukturen til RNA ved forskjellige temperaturer, og bruk mFold online-verktøyet for å bestemme den sekundære strukturen til målfragmentet under spesifikke temperatur- og saltkonsentrasjonsforhold.Ved 55 °C er den sekundære strukturen til RNA fortsatt svært kompleks, revers transkriptase kan ikke fungere, og den sekundære strukturen kan ikke løses fullstendig før 65 °C, mens den optimale temperaturen til AMV og M-MLV er langt lavere enn denne temperaturen.
hva å gjøre?Den sekundære strukturen er den komplementære sammenkoblingen av selve malen, noe som fører til sterk konkurranse mellom primeren og revers transkriptase og malen, noe som resulterer i en rekke problemer som lav E og dårlig repeterbarhet.
hva å gjøre?Øk kun glødetemperaturen så mye som mulig.
Mange reagensprodusenter forbedrer revers transkriptase gjennom genteknologi.Noen øker reaksjonstemperaturen, slik som Jifan og Aidelai, og noen fjerner den aktive gruppen av RNase H-enzymet for å forbedre affiniteten mellom enzymet og RNA-malen.Høy affinitet kan konkurransedyktig presse ut den sekundære strukturen og lese gjennom jevnt, og også i stor grad forbedre effektiviteten til omvendt transkripsjon.
Nøkkelpunkt: Revers transkripsjon er viktigere for å etterstrebe konsistensen av revers transkripsjonseffektivitet (enzymer må ikke bare være effektive, men også stabile), i stedet for mengden prøve som er lastet, hvis det ikke er en spesielt storskala fluorescerende kvantitativ PCR, vil det ikke være mulig i det hele tatt.Flere cDNAer.
Ulike produsenter har også gjort en viss innsats i jakten på konsistens.For eksempel har de fleste selskaper nå pakket omvendt transkripsjon som et standardsett for salg, noe som er et godt valg.
For eksempel Foregenes RT Easy Series-sett:
RT Easy I (Master premix for første streng cDNA syntesesett)
MIQE (5) – målgeninformasjon

Figuren ovenfor forklarer
1. Hvorvidt dette genet er effektivt for gjentatte eksperimenter kan generelt verifiseres ved gjentatte eksperimenter.
2. Gen-ID, vet du.
3. Genlengde, den totale lengden på målgenet er definitivt ikke noe problem.Når du designer primere, sørg for at lengden på amplikonet er mellom 80-200bp for å sikre en bedre amplifikasjonseffektivitet.
4. Sekvens Blast-sammenligningsinformasjon, målgenet må sammenlignes i genbanken for å forhindre uspesifikk amplifikasjon.
5. Tilstedeværelse av pseudogener.Et pseudogen er en DNA-sekvens som ligner på et normalt gen, men mister sin normale funksjon.Det eksisterer ofte i multi-genfamilien av eukaryoter.Det er vanligvis representert med ψ.Det er en ikke-funksjonell genomisk DNA-kopi i genomet som er veldig lik den kodende gensekvensen., er generelt ikke transkribert, og har ingen klar fysiologisk betydning.
6. Plassering av primere i forhold til eksoner og introner.I de første årene, da vi løste problemet med DNA-kontaminering, tok vi ofte hensyn til posisjonene til primere, eksoner og introner, og vurderte generelt å designe primere på tvers av introner for å unngå DNA-amplifisering.Se figuren nedenfor: svart representerer introner, forskjellige blåtoner representerer eksoner, rosa representerer vanlige primere, og knallrødt representerer intronspennende primere.
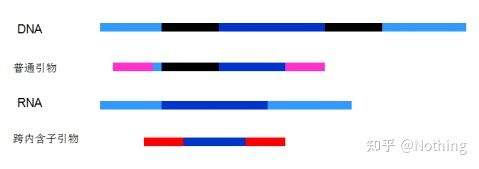
Skjematisk, aldri sant
For en perfekt plan dette virker, men i de fleste tilfeller er ikke trans-intron-primerne så magiske som forestilt, og de vil også forårsake uspesifikk amplifikasjon.Så den beste måten å forhindre DNA-kontaminering på er å fjerne DNA fullstendig.
7. Konformasjonsprediksjon.Bruk dette eksemplet igjen, bruk mFold online-verktøyet for å bestemme den sekundære strukturen til målfragmentet ved en spesifikk temperatur og saltkonsentrasjon.
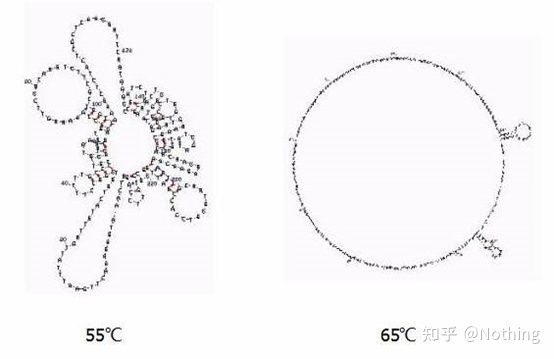
Sekundær struktur av RNA ved forskjellige temperaturer
Den sekundære strukturen er den komplementære sammenkoblingen av selve malen, noe som vil føre til sterk konkurranse mellom primer- og malparing, og sjansene for primerbinding er mindre, noe som resulterer i en rekke problemer som lav E og dårlig repeterbarhet.Gjennom programvareprediksjon, hvis det ikke er noe sekundært strukturproblem, ville det vært flott.Hvis det er det, vil oppfølgingsartikkelen vår spesifikt diskutere hvordan du løser dette problemet.
MIQE (6)—qPCR-oligonukleotider

For fluorescerende kvantitativ PCR er det første du sliter med hver dag RNA-ekstraksjon, og det andre kan være primerdesign.
Først og fremst sjekker vi fortsatt reglene om primerdesign i henhold til MIQE-sjekklisten.Det er så enkelt at skurkene kan le, og vi kan fullføre det i én setning: finn ut sekvensen og posisjonen til primerproben og modifikasjonsmetoden.For primerrensemetoden er primersyntese så billig for tiden, qPCR er verdig PAGE og høyere rensemetoder, og informasjonen til synteseinstrumentet er ikke viktig.Mange har drevet med primere i flere tiår og vet ikke at synthesizeren er ABI3900.
Når det gjelder prinsippene for primerdesign, trenger du ikke å huske dem utenat, fordi de fleste primerdesignprogramvare eller nettverktøy kan ta seg av disse problemene (anbefalt nettverktøy primer3.ut.ee/), og 99,999 % av primerdesign gjøres ikke manuelt Se, forfatteren designer noen ganger hundrevis av primere om dagen, hvis du leser.
Bare sjekk følgende punkter etter at primerne er designet:
1. Designprimere nær 3'-enden: Ved bruk av oligo dT-primere for cDNA-første-trådsyntese, med tanke på revers transkripsjonseffektiviteten og RNA-integriteten, må de utformede primerne utformes nær 3'-enden for å forbedre amplifikasjonseffektiviteten.Bruk et bilde for å forklare som følger (det er ingen måte å forstå dette på):
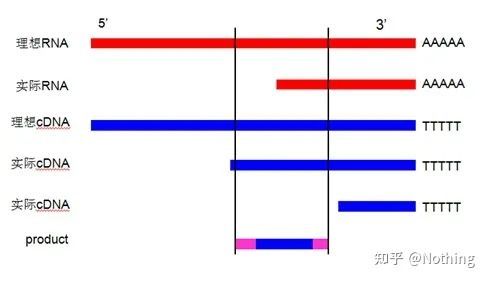
Hvorfor skal primere designes nær 3′-enden, det må ikke være sant
2. TM-verdi: Tm-verdien er ved 55-65°C (fordi eksonukleaseaktiviteten er høyest ved 60°C), og GC-innholdet er på 40%-60%.
3. BLAST: For å unngå uspesifikk amplifikasjon av genomet, må Blast brukes til supplerende verifikasjon.
MIQE(7)—qPCR-prosess
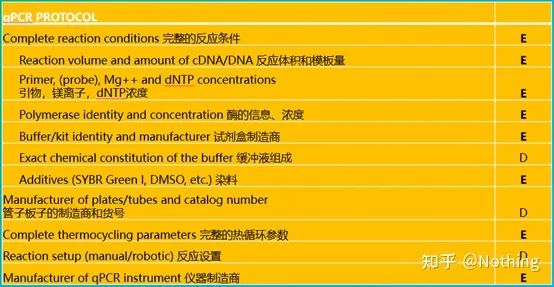
1. qPCR-sett
I henhold til kravene til MIQE, må vi tydelig beskrive de fullstendige reaksjonsbetingelsene i artikkelen, inkludert konfigurasjonen av PCR-reaksjonssystemet, hvilket sett som brukes, hvem er produsenten, hvor stort er reaksjonssystemet, om fargemetoden eller probemetoden brukes, PCR-programinnstillinger.Veteransjåfører vil definitivt finne at så lenge settet er valgt, er informasjonen ovenfor i utgangspunktet bestemt.
For tiden er produksjon og produksjon av fluorescerende kvantitative PCR-sett en svært moden teknologi.Så lenge du ikke velger ekstremt dårlige produsenter, er sannsynligheten for problemer ikke stor, men vi ønsker likevel å dele noen punkter med deg:
Hot-start Taq enzym:Den viktigste delen av PCR er hot-start Taq enzymet.Hot-start enzymene på markedet er generelt delt inn i to typer, den ene er et kjemisk modifisert hot-start enzym (du kan tenke deg det som parafininnstøping), og den andre er Er et hot-start enzym for antistoffmodifisering (antigen-antistoffbinding).Kjemisk modifikasjon er en tidlig måte å varmstarte enzymer på.Når en viss temperatur er nådd, vil enzymet frigjøre sin aktivitet.Det antistoffmodifiserte hot-start enzymet bruker biologiske metoder for å blokkere aktiviteten til enzymet.Når en viss temperatur er nådd, vil antistoffet denatureres og inaktiveres som et protein, og enzymaktiviteten bringes i spill.
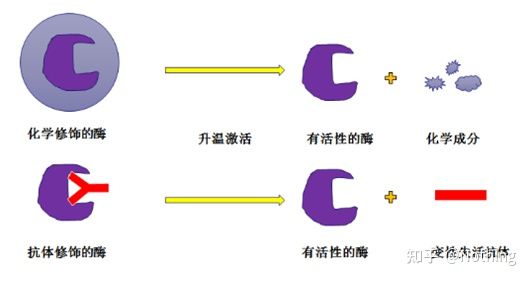
Men hva er nytten med dette?Dette er tilfellet, frigjøringsaktiviteten til antistoffmodifiserte enzymer er raskere enn kjemisk modifiserte enzymer, så med tanke på sensitivitet har antistoffmodifiserte enzymer en liten fordel, slik at det i utgangspunktet ikke er noen kjemisk modifiserte enzymer i settene på markedet.Hvis det er det, er denne produsentens teknologi fortsatt fast i årtusenet.
Magnesiumionkonsentrasjon:Magnesiumionkonsentrasjon er svært viktig i PCR-reaksjonen.Passende magnesiumionekonsentrasjon kan fremme frigjøringen av Taq-enzymaktivitet.Hvis konsentrasjonen er for lav, vil enzymaktiviteten reduseres betydelig;hvis konsentrasjonen er for høy, vil den enzymkatalyserte ikke-spesifikke amplifikasjonen bli forsterket.Konsentrasjonen av magnesiumioner vil også påvirke annealingen av primere, smeltetemperaturen til malen og PCR-produkter, og dermed påvirke utbyttet av amplifiserte fragmenter.Konsentrasjonen av magnesiumioner er generelt kontrollert til 25 mM.Selvfølgelig, for et godt sett, må konsentrasjonen av magnesiumioner være godt kontrollert.Noen forhandlere legger til et magnesiumion-chelateringsmiddel til reagenset, som kan oppnå effekten av automatisk justering av magnesiumionekonsentrasjonen.
Fluorescerende fargestoffkonsentrasjon:Fluorescerende fargestoff, som er SYBR Green vi vanligvis bruker, genererer hovedsakelig fluorescens ved å binde seg til den mindre rillen av dobbelttrådet DNA, fordi bindingen av fargestoffet til dobbelttrådet DNA er uspesifikk, det vil si så lenge dobbelttrådet DNA er kombinert med det, kan fluorescens oppstå, så vil primer-dimerer kombineres med bakgrunnsmaler i systemet.
PS: På grunn av de lysfølsomme egenskapene er produkter på markedet generelt pakket i brune ugjennomsiktige sentrifugerør (som vist på bildet nedenfor).Dette vil imidlertid støte på et problem.Det er vanskelig å se om væsken suges ved prøvetaking.I så måte er Qingke faktisk den mest brukervennlige (som vist på bildet nedenfor), og det gjennomsiktige røret er pakket i en ugjennomsiktig blikkpose.Legg den deretter i en blikkpose, med tanke på bekvemmeligheten av å unngå lys og prøvetaking.Du må velge riktig produktnummer.TSE204 er en super kostnadseffektiv tilværelse, som gjør at jeg får lyst til å plante gress.

Konsentrasjonen av det fluorescerende fargestoffet er også veldig viktig.Hvis konsentrasjonen er for lav, vil ikke amplifikasjonskurven gå opp i det senere stadiet og er ikke perfekt;hvis konsentrasjonen er for høy, vil det forårsake støyinterferens.Siden den fluorescerende kvantitative PCR hovedsakelig avhenger av CT-verdien, hvis konsentrasjonen av det fluorescerende fargestoffet ikke er riktig justert, er lavpunktet bedre enn høydepunktet.Selvfølgelig er riktig fargestoffkonsentrasjon den beste.
ROX: ROX-fargestoffer brukes til å korrigere for fluorescenssignalfeil fra brønn til brønn.Noen instrumentprodusenter krever kalibrering, mens andre ikke gjør det.For eksempel krever bruk av Thermo Fisher Scientifics sanntids PCR-amplifikasjonsinstrument vanligvis kalibrering, inkludert 7300, 7500, 7500Fast, StepOnePlus osv. De generelle settinstruksjonene vil beskrive det.
Foregenes qPCR Mix inneholder også ROX-fargestoff, som er praktisk å bruke i ulike modeller.
Svak hydrogenbindingsbehandling: Behandling av svake hydrogenbindinger er en relativt teknisk sak.Ingenting har lest manualene til mange sett, men ingen av dem nevnte dette emnet.Faktisk er det så viktig.Kombinasjonen av baser avhenger hovedsakelig av styrken til hydrogenbindinger.Sterke hydrogenbindinger er normal amplifikasjon, og svake hydrogenbindinger fører til uspesifikk amplifikasjon.Hvis svake hydrogenbindinger ikke kan elimineres godt, kan ikke uspesifikk amplifikasjon unngås.Innenfor forfatterens omfang er det bare noen få selskaper som har lagt merke til dette problemet.Når du kjøper settet kan du henvise til om du har vurdert en løsning i denne forbindelse for det settet du ønsker å velge.
Reaksjonsvolum: 20-50ul-systemet er mer vanlig brukt, og mindre volumer vil sannsynligvis forårsake feil.Generelt sett vil kitinstruksjonene anbefale bruk av PCR-reaksjonsvolumer.Ikke vær smart og bruk mindre volumer for å spare kostnader.målet om.Volumet anbefalt av kjøpmennene er faktisk testet, og det kan være at de ikke kan løse problemet med feil forårsaket av små volumer.
2. Produsent og artikkelnummer på rørplaten
Alle kjenner til prinsippet om fluorescerende kvantitativ PCR.Fluorescensinnsamling utføres hovedsakelig gjennom PCR-rørhetter.Når du velger PCR-forbruksvarer, vær oppmerksom på to punkter: god lysgjennomgang og egnet for instrumentet.Generelt sett er brettene og rørene til mainstream-merker fine, men du må velge nøye med tanke på tilpasning, ellers vil du ikke kunne bruke instrumentet.

4. Kunnskap på toppnivå
MIQE (8)—qPCR-validering
Dette er toppprioriteten til qPCR!Så mange helter har falt i sanden her.Det er selvfølgelig også mulig at du er heldig og genene du studerte er enkle, så du fløt gjennom ishulen langs vinden.Verifikasjonsinformasjonen til qPCR er ment å teste påliteligheten til dataene.Vi viser den nødvendige bekreftelsesinformasjonen som følger:
1. Spesifisitetstest
Spesifisiteten til målgenamplifikasjon testes ved å sjekke om elektroforesebildet er et enkelt bånd;sekvensering verifisering;smeltekurve for å se om toppkartet er enkelt;enzymfordøyelsesverifisering og andre metoder.
Her fokuserer vi på tanalysen av ikke-spesifikk amplifikasjon ved bruk av metoden for smeltekurver.Generelt sett, når vi designer primere, kreves det at størrelsen på produktfragmentet er i området 80-200 bp, noe som gjør at smeltetemperaturen til PCR-produktet er 80-85 °C.Derfor, hvis det er diverse topper, må det være andre ikke-spesifikke amplifikasjonsprodukter;hvis toppen viser seg under 80°C, anses den generelt for å være en primerdimer;hvis toppen vises over 85°C, anses det generelt for å være DNA-kontaminering eller mer uspesifikk amplifikasjon av store fragmenter.
Merk: Noen ganger er det bare en enkelt topp ved 80°C.På dette tidspunktet må dette konseptet følges.Det er sannsynlig at amplifikasjonsresultatene alle er primerdimerer.
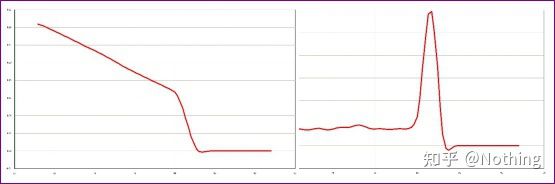
Normal smeltekurve (enkelt topp uten uspesifikk forsterkning)
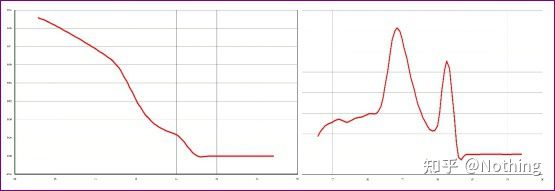
Problematisk smeltekurve (ikke-spesifikk forsterkning av falske topper)
【Kasusanalyse】
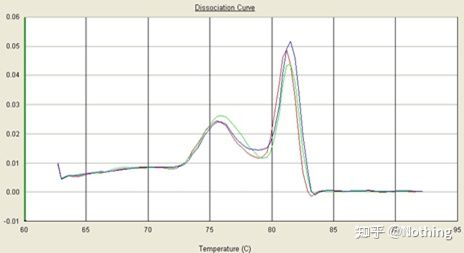
Det er en hovedtopp, men primer-dimeren er alvorlig
Enkeltoppssmeltekurven i figuren nedenfor kan lett lure øynene dine, og tro at det er et perfekt eksperiment, men resultatet er helt feil.På dette tidspunktet må vi se på smeltetemperaturen.Topptemperaturen er under 80°C, som er fullstendig primer-dimer.
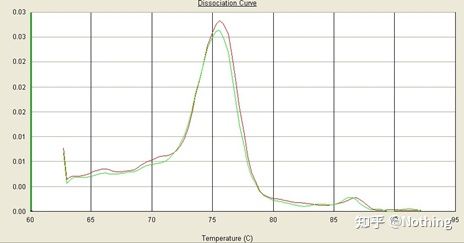
Ingen målfragment, alle primer-dimerer
Her kan ikke broren min stoppe.Bildet under er et bilde tatt med en mobiltelefon sendt til meg av en drittsekk.Reagensene han brukte er alle vanlig brukte merker i bransjen.Han endret fra ett T-prefiksmerke til et annet T-prefiksmerke.Jeg tror du allerede har gjettet det.Skumpet ropte til meg: «Reagensen som ble brukt på det første bildet er for bra, og toppen er enkel.Senere, etter å ha brukt reagenset du anbefalte, blir det som det andre bildet, med blandede topper.Du har gjort meg ulykkelig."
Skille de to grafene.Ved første øyekast har den ene en enkelt topp, og den andre har en dobbel topp.Tull, en enkelt topp er selvfølgelig greit.Er det sant?
Verre enn Dou E, hvis jeg legger de to bildene på bildet under, vil du forstå umiddelbart.Faktisk blir vi lett lammet av denne typen bilder.Etter nøye analyse fant vi at: toppen av den første figuren er ved 75°C, som er fullstendig primerdimer;toppen av den andre figuren vises ved 75°C og 82°C, i det minste er det Produktet vises.
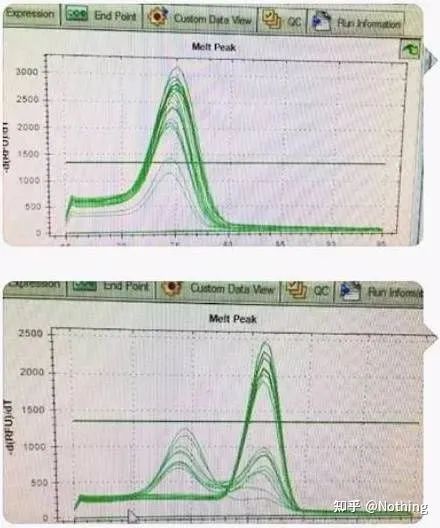
Bilder av tilbakemeldinger fra studenter
Så det grunnleggende problemet er ikke problemet med reagenser, men problemet med primerdesign.Samtidig beviser det også at noen store merker ikke er av jernkvalitet, og det beviser også det broren min sa før: Det er ikke reagensmerket som støtter artikkelen din.Det er artikkelen din som støttet opp merket av reagenser.Tenk deg, hvis skurken ikke endret reagensene, ville feil data bli sendt til journalen, og det som ville skje ville være en tragedie.
2. Ct-verdi for blankkontroll
Ikke forklar, hvis blankkontrollen har en Ct-verdi, er det ikke forurensning?Du må imidlertid fortsatt forstå hvilken blank kontroll som har en Ct-verdi.Hvis det er NTC, betyr det at det er fremmed DNA som reagenskontaminering.Hvis det er NRT, betyr det at det ekstraherte RNA har DNA-kontaminering.
3. Standardkurve
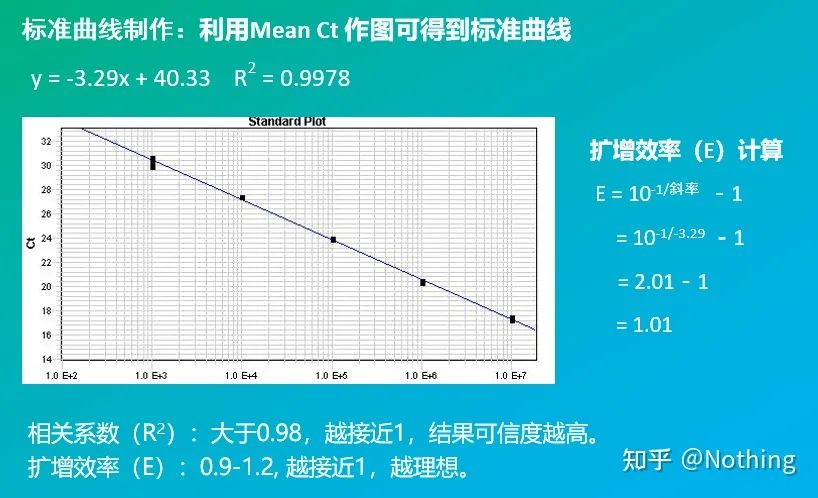
Inkludert helnings- og beregningsformelen, kan PCR-effektiviteten beregnes gjennom formelen.Et perfekt eksperiment krever at helningen til standardkurven nærmer seg 3,32, og at R² nærmer seg 0,9999.
4. Lineært dynamisk område
Det dynamiske området for reaksjonen er lineært.I henhold til malen som brukes til å generere standardkurven, bør det dynamiske området inkludere minst 5 konsentrasjonsgradienter, og ta hensyn til endringen av Ct-verdier ved høye konsentrasjonsgradienter og lave konsentrasjonsgradienter.
5. Deteksjonsnøyaktighet
Endringer i qPCR-resultater, det vil si dårlig repeterbarhet, det vil si dårlig presisjon, er forårsaket av mange faktorer, inkludert temperatur, konsentrasjon og drift.qPCR-presisjonen blir generelt mindre kontrollerbar ettersom antall kopier reduseres.Ideelt sett innenfor eksperimentell variasjon, bør denne tekniske variasjonen være forskjellig fra biologisk variasjon, og biologiske replikater kan direkte adressere statistiske forskjeller i qPCR-resultater mellom grupper eller behandlinger.Spesielt for diagnostiske analyser må den beste presisjonen (repeterbarhet) mellom analysene på tvers av steder og operatører rapporteres.
6. Deteksjonseffektivitet og LOD (i multipleks qPCR)
LOD er den laveste konsentrasjonen på 95 % av positive prøver påvist.Med andre ord, konsentrasjonen av LOD inneholdt i et sett med målgenreplikater bør ikke overstige 5 % av de mislykkede reaksjonene.Når du gjør multipleks qPCR-analyse, spesielt for samtidig påvisning av punktmutasjoner eller polymorfismer, må multipleks qPCR gi bevis for at nøyaktigheten til flere målfragmenter ikke er kompromittert i samme rør, multippel deteksjon og enkeltrørdeteksjon Effektivitet og LOD skal være den samme.Spesielt når høykonsentrasjonsmålgener og lavkonsentrasjonsmålgener amplifiseres samtidig, må dette problemet tas hensyn til.
Problemer og løsningerGenerelt sett fokuserer problemene som ofte oppstår i qPCR-feilsøking på følgende aspekter:
·uspesifikk amplifikasjon
·Vanskelig valg av primerkonsentrasjon og problemer med primer-dimere
·Glødetemperaturen er unøyaktig
·Sekundær struktur påvirker forsterkningseffektiviteten
uspesifikk amplifikasjon
ikke-spesifikk forsterkningoppstår, vurderes det generelt om primerdesignet ikke er egnet, men hvis du ikke har det travelt med å bytte primer, kan du prøve følgende metoder først (prinsippet er også vedlagt):
· Øk utglødningstemperaturen – prøv å gjøre svake hydrogenbindinger ute av stand til å opprettholde;
· Forkort gløde- og forlengelsestid – reduser sjansen for svake hydrogenbindinger;
· Reduser primerkonsentrasjonen – reduser sjansen for binding av overflødige primere og ikke-målområder;
Lav forsterkningseffektivitet
Den motsatte situasjonen til ikke-spesifikk forsterkning – lav forsterkningseffektivitet, og tiltakene for å håndtere lav forsterkningseffektivitet er akkurat det motsatte:
· Forleng gløde- og forlengelsestiden;
· Bytt til tre-trinns PCR og reduser utglødningstemperaturen;
· Øk primerkonsentrasjonen;
Ps: Mange doktorgradsstudenter født på 90-tallet er uvillige til å studere hvordan man feilsøker eksperimenter, og håper at settet kan løse problemet fullstendig (hvis du vil gå til et reagensfirma for å gjøre forskning og utvikling etter endt utdanning), faktisk tenker reagensprodusentene også på denne måten, jeg håper det er en idiot. absorpsjonsfaktorer.For å enkelt løse problemet, må idioter fortsatt lese introduksjonen til reagensfirmaet for å se om det er en faktor som absorberer svake hydrogenbindinger.
Vanskelig valg av primerkonsentrasjon og problemer med primer-dimere
Metode 1: Generelt sett har settinstruksjonene for qPCR anbefalte systemer og anbefalte primerkonsentrasjoner.
Metode 2: Feilsøking ved å sette primerkonsentrasjonsgradienten.Bildet under er stjålet fra et selskap for å illustrere.Figuren nedenfor viser de kvantitative fluorescensresultatene laget med tre primerkonsentrasjonsgradienter (100nM, 250nM, 500nM) og fire malkonsentrasjonsgradienter (0,1ng, 1ng, 10ng, 100ng).Ct-verdien til de eksperimentelle resultatene er plottet som følger:
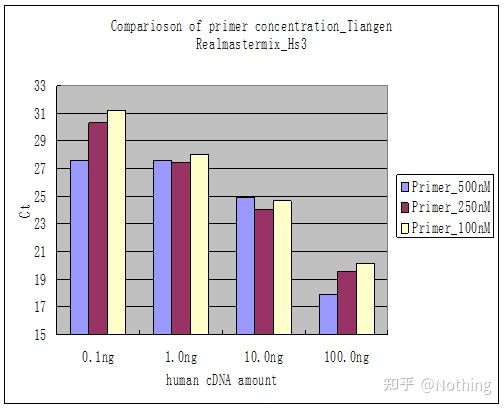
Valg av primerkonsentrasjon Konkatener hver primerkonsentrasjon i en linje som følger:
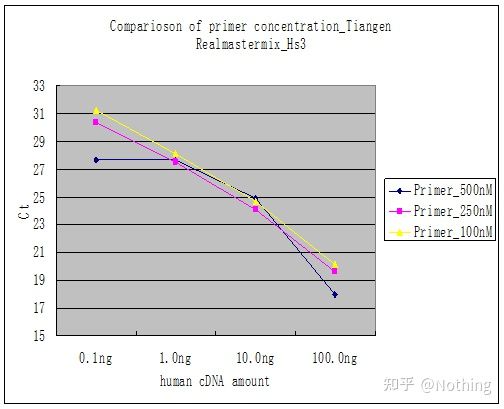
Valget av primerkonsentrasjon er åpenbart, det lineære forholdet mellom primerkonsentrasjonen på 100nM og 250nM er bedre, og det lineære forholdet til primerkonsentrasjonen på 500nM er relativt dårlig.I 100nM og 250nM er Ct-verdien på 250nM relativt liten, så den optimale primerkonsentrasjonen er 250nM.Generelt kan alvorlige primer-dimerer sees i smeltekurven.Hva om de utformede primerne ikke kan unngå primer-dimerer?
Metode 3: Reduser mengden primere og øk annealingstemperaturen (ingen grunn til å forklare).
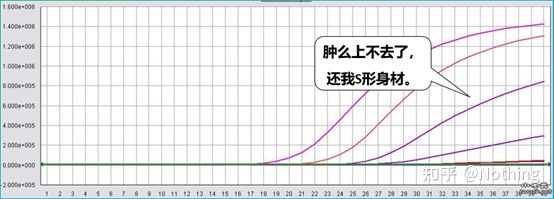
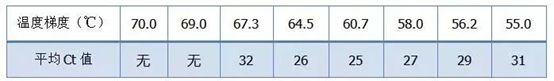
Den empiriske verdien av glødetemperaturen er 60°C.Hvis du ikke er sikker, hvordan velge en mer passende glødetemperatur?Svaret er det samme som valg av primerkonsentrasjon –gradienttest.Ta et bilde fra Bio-rad-selskapet for å illustrere problemet.For amplifisering av et bestemt målfragment, sett åtte temperaturgradienter, hver med tre repetisjoner, og den oppnådde amplifikasjonskurven er som følger:
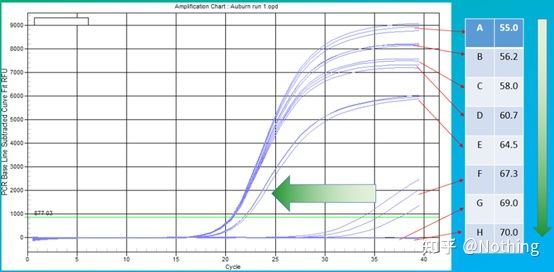
valg av glødetemperatur:
·70°C, 69°C—I utgangspunktet kan ikke primerne kombineres, så det er ingen amplifikasjon.
·67,3°C – Det er en liten mengde forsterkning i begynnelsen, og Ct-verdien er relativt stor.
·64,5°C——Ct-verdien synker.
·Ved 60,7°C, 58,0°C, 56,2°C og 55,0°C hadde Ct-verdiene i utgangspunktet en tendens til å være stabile, men de endelige fluorescensverdiene var forskjellige.
Hvordan velge?Prinsipp: Det første prinsippet er den høyere Ct-verdien.For samme Ct-verdi, velg en høyere utglødningstemperatur for å unngå dimerisering og uspesifikk amplifikasjon.Selv om det er en høyere fluorescensverdi ved 55°C, kan det være dimerer eller ikke-spesifikk amplifikasjon i den.
Men hvis du er like smart som deg, vil du definitivt tenke: Logisk sett, hvis PCR-reaksjonen er veldig spesifikk, så lenge primerkonsentrasjonen overstiger minimumskravet, skal høye og lave punkter ikke ha noen effekt, akkurat som fluorescerende fargestoffer og dNTP.Faktisk, så lenge annealingstemperaturen er optimalisert riktig, vil effekten av primerkonsentrasjon på Ct-verdien naturligvis bli minimalisert.
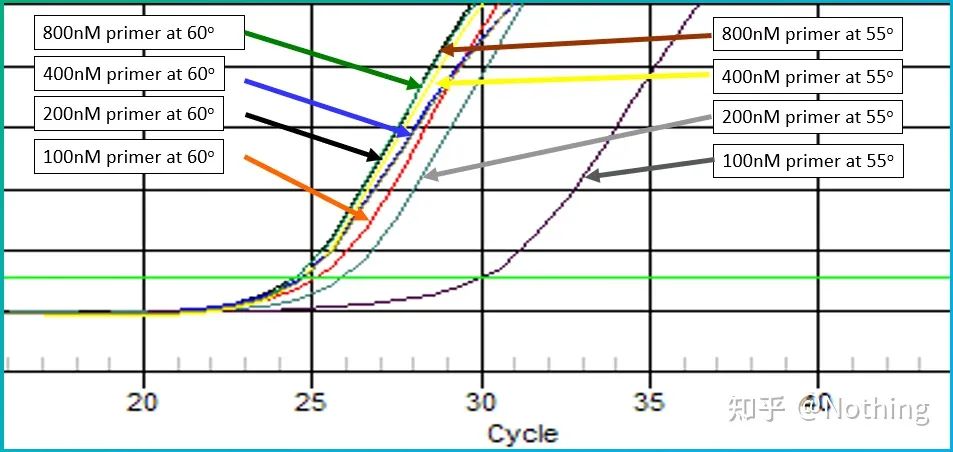
Glødetemperaturen er optimalisert riktig, og effekten av primerkonsentrasjon på CT vil bli minimalisert
Sekundær struktur påvirker amplifikasjonseffektiviteten
La oss ta bildet fra Bio-rad for å illustrere problemet.Den designer også en temperaturgradient for å forsterke et gen med en sekundær struktur.
Sekundær struktur oppstår
Det kan sees at når temperaturgradienten avtar, begynner produktene å dukke opp og Ct-verdien beveger seg fremover, når minimumsverdien ved 60,7°C, og når temperaturgradienten avtar, blir Ct-verdien større.Motsatt, når temperaturen øker, åpnes sekundærstrukturen og forsterkningseffektiviteten øker.Etter å ha nådd en viss temperatur kan ikke økning av temperaturen forbedre forsterkningseffektiviteten.Fordi primerne ikke kan kombineres stabilt på dette tidspunktet.Derfor,se etter temperaturen med den laveste Ct-verdien, som er den beste temperaturen for å forsterke den sekundære strukturmalen!Selvfølgelig må smarte idioter vite at hvis det ikke er nødvendig, er det best å endre primerne og unngå den sekundære strukturregionen.
5. Søknadsnivå
MIQE—Dataanalyse

Dataanalysen er hovedsakelig gitt av det fluorescerende kvantitative PCR-instrumentet.I forrige artikkel er det gjort mye dataanalysearbeid, som for eksempel blankkontrollen, som er forklart i utformingen av eksperimentet.De interne referansegenene, repetisjonstall osv. er avklart., her forklarer vi hovedsakelig bruken av qPCR.
qPCR er mye brukt, og eksperimentell verifikasjon og nukleinsyrediagnose er de mest brukte scenariene.
absolutt kvantifisering
Logg (startkonsentrasjon) har en lineær sammenheng med antall sykluser.En standardkurve kan trekkes fra en standard med kjent initialt kopiantall, det vil si at det lineære forholdet til amplifikasjonsreaksjonen kan oppnås.I henhold til Ct-verdien til prøven kan konsentrasjonen i prøven beregnes.Antall maler som skal inkluderes.
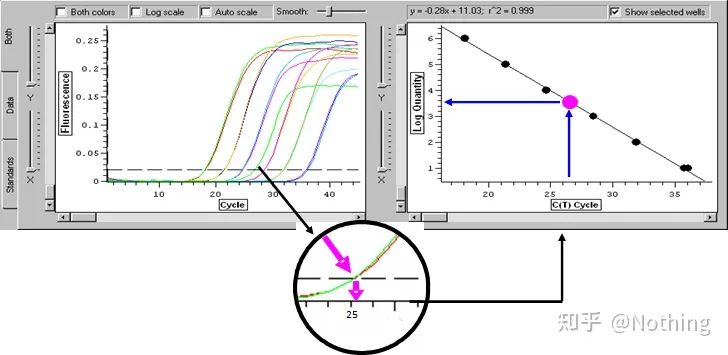
Absolutt kvantitativ beregningsmetode
Absolutt kvantifisering må baseres på standardkurven.For å lage en standardkurve kreves det en standard.Vanligvis er standarden et plasmid oppnådd ved å klone målgenet.Hvorfor er det et plasmid?Fordi sirkulært plasmid DNA er det mest stabile.Fortynn standardproduktet i 5 til 6 gradienter i henhold til doblingsforholdet (10 ganger fortynning), og vær oppmerksom på jevnheten ved fortynning.La Ct-verdien falle mellom 15-30.
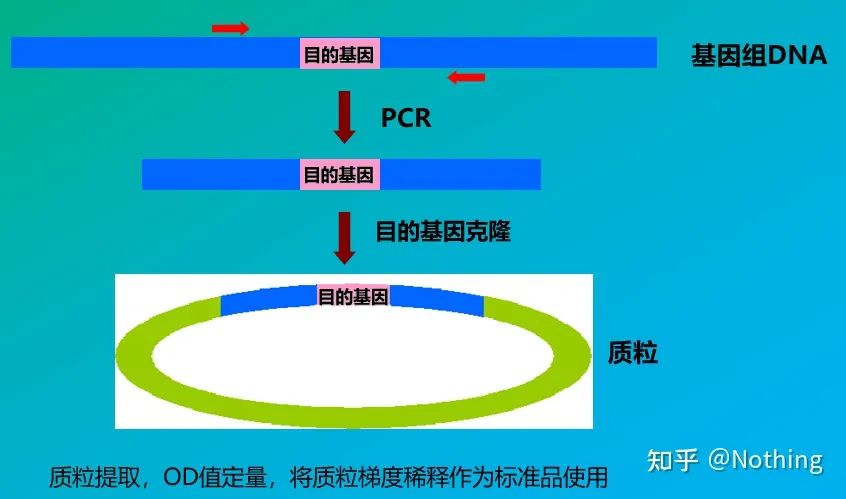
Standard forberedelse
Samtidig bør prøven som skal testes også fortynnes tilsvarende (husk fortynningsfaktoren), og Ct-verdien bør også falle mellom 15-30.Standardproduktet + prøven som skal testes settes på maskinen sammen.Etter kjøringen ble det laget en standardkurve med standardstoffet, og prøvene som skulle testes ble brakt inn i standardkurven for å beregne konsentrasjonen.
Hepatitt B-virus HBV kvantifisering er en typisk absolutt kvantifisering, som kan beregne viruskopitallet i 1 ml blod.
Beregning av kopinummer
Prøvekonsentrasjon som skal testes (ng/ul) = OD260 × 50 ug/ml × fortynningsfaktor
Prøvens molekylvekt = antall baser × 324
Kopinummeret til prøven som skal testes (kopier/ul) = konsentrasjonen av prøven som skal testes / molekylvekten til prøven × 6 × 1014

Beregningsmetode for kopinummer
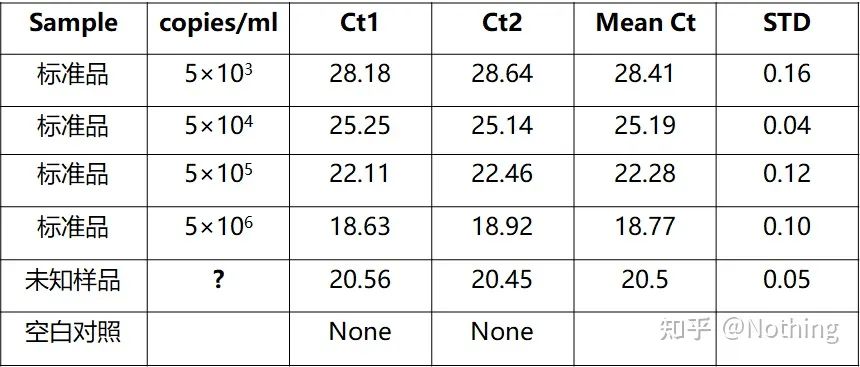

Ovenstående er beregningsmetoden for å bestemme mengden.Dette er et matematisk problem som kan løses etter endt ungdomsskole, og matematiske problemer løses vanligvis av datamaskiner.Hvis du ikke forstår, kan du komme for å kommunisere.
relativ kvantifisering
Relativ kvantifisering brukes hovedsakelig i vitenskapelig forskning.Hvor mange virus er det i 1 ml blod, og det er et DNA-virus, dette er en relativt deterministisk hendelse: mengden blod kan bestemmes, og DNA-viruset er relativt stabilt.Imidlertid er det vanskelig for oss å sammenligne antall transkripsjonskopier av et bestemt gen i et blad, fordi det er vanskelig å bestemme størrelsen, vekten og ømheten til bladet, mengden ekstrahert RNA er vanskelig å bestemme, og effektiviteten til omvendt transkripsjon er også vanskelig å bestemme, det vil si at ethvert trinn kan gjøre at eksperimentelle data har feil og ikke kan brukes.
Derfor må relativ kvantifisering introdusere et element:det interne referansegenet.
Med andre ord er relativ kvantifisering faktisk en sammenligning mellom målgenet og det interne referansegenet.Sammenlignet i samme vev og samme celle er påvirkningen av prøvestørrelse, RNA-ekstraksjonsmengde, revers transkripsjonseffektivitet og PCR-effektivitet relativt liten.På grunn av den lille prøvestørrelsen ble både interne referansegener og målgener relativt redusert.Dette er grunnen til at vi har lagt vekt på enhetlighet og stabilitet før.
Interne referansegener er generelthusholdningsgener(husholdende gener), som refererer til en klasse av gener som er stabilt uttrykt i alle celler, og deres produkter er nødvendige for å opprettholde cellenes grunnleggende livsaktiviteter.
Ikke forveksle dette konseptet.Husholdningsgener er biologiske funksjonsbegreper, mens interne referansegener er eksperimentelle faguttrykk.Husholdningsgener må bestå validering før de kan velges som interne referansegener.
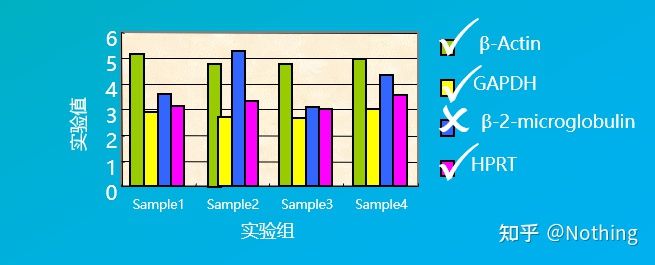
For eksempel valgte vi flere husholdningsgener i figuren nedenfor for å teste ekspresjonsnivåene deres i forskjellige vevsceller, og fant at ekspresjonsnivåene til β-2-mikroglobulin var ganske forskjellige fra de andre tre genene, så de kunne ikke brukes som interne referansegener.
Etter å ha forstått korreksjonsfunksjonen til det interne referansegenet, utledes to algoritmer på grunn av introduksjonen av det interne referansegenet.
·dobbel standard kurvemetode
·2 – △△Ct-metode (CT-verdisammenligningsmetode)
Hvis du er interessert i å studere arter og genfunksjoner, vennligst gi opp forskningen på algoritmer og bruk formler direkte, eller bruk maskiner direkte;hvis du er en straight fyr i matematikk og ingeniørfag, vær så snill.
dobbel standard kurvemetode
Kvantifiser målgenet og husholdningsgenet til kontrollprøven og prøven som skal testes gjennom standardkurven, og beregn deretter den relative verdien i henhold til beregningsformelen, som er det relative uttrykksnivået.
Fordeler: enkel analyse, relativt enkel eksperimentell optimalisering
Ulempe: For hvert gen må hver runde med eksperimenter lage en standardkurve
Anvendelse: En av de to mest brukte og anerkjente relative kvantitative metodene i studiet av genekspresjonsregulering
Formelen er som følger:

Eksempler er som følger:
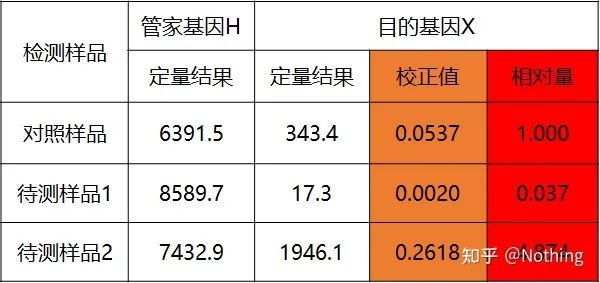
Beregn den relative mengden basert på det kvantitative resultatet
2 – △△Ct-metode (CT-verdisammenligningsmetode)

Fordeler: Ingen grunn til å lage en standardkurve
Ulemper: Det antas at amplifikasjonseffektiviteten er nær 100 %;standardavviket er < 5 %, og standardkurven og effektiviteten mellom hver amplifikasjon antas å være konsistente;optimering av eksperimentelle forhold er mer komplisert.
Anvendelse: En av de to mest brukte og anerkjente relative kvantitative metodene i studiet av genekspresjonsregulering
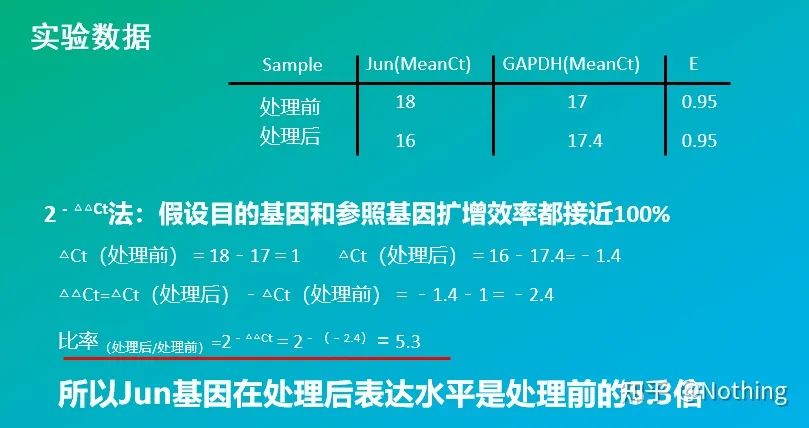
Selvfølgelig er amplifikasjonseffektiviteten vanligvis umulig å være perfekt 1. Korreksjonsmetode: Hvis vi vet at målgenet og referansegenet har samme amplifikasjonseffektivitet, men amplifikasjonseffektiviteten er ikke lik 1, så kan 2-△△Ct korrigeres som: (1+E )-△△Ct, kan for eksempel 0-9-formelen korrigeres til 5. .95–△△Ct
Så langt har innholdet om fluorescerende kvantitativ PCR tatt slutt.
Innleggstid: Apr-06-2023



