



Som ny i laboratoriet er det ingen god jobb å sile ut positive planter fra en haug med planter med lav konverteringsrate.For det første må DNA ekstraheres fra et stort antall prøver én etter én, og deretter vil de fremmede genene bli påvist ved PCR.Resultatene er imidlertid ofte blanke og bånd med noen få gjenstander av og til, men det er umulig å avgjøre om det er tapte eller falske deteksjoner..Er det veldig hjelpeløst å møte slike eksperimentelle prosesser og resultater?Ikke bekymre deg, bror lærer deg hvordan du enkelt og nøyaktig siler ut transgene positive planter.
Trinn 1
Design deteksjonsprimere

Bestem det endogene genet og det eksogene genet som skal påvises i henhold til prøven som skal testes, og velg en representativ 100-500bp sekvens i genet for primerdesign.Gode primere kan sikre nøyaktigheten av deteksjonsresultatene og forkorte deteksjonstiden (se vedlegget for vanlig brukte deteksjonsprimere).
Merk: De nydesignede primerne må optimalisere reaksjonsforholdene og verifisere nøyaktigheten, presisjonen og deteksjonsgrensen for deteksjonen før deteksjon i stor skala.
Steg 2
Design eksperimentell protokoll

Positiv kontroll: Bruk det rensede DNA-et som inneholder målfragmentet som en mal for å bestemme om PCR-reaksjonssystemet og -forholdene er normale.
Negativ/blank kontroll: Bruk DNA-malen eller ddH2O som ikke inneholder målfragmentet som mal for å oppdage om det er en kontaminasjonskilde i PCR-systemet.
Intern referansekontroll: bruk primer/probe-kombinasjonen av det endogene genet til prøven som skal testes for å evaluere om malen kan påvises ved PCR.
Legge merke til:
Positive, negative/blanke kontroller og internkontrollkontroller bør angis for hver test for å evaluere gyldigheten av de eksperimentelle resultatene.
Eksperimentforberedelse

Før bruk, sjekk om løsningen er jevnt blandet.Hvis det oppdages nedbør, må det løses opp og blandes i henhold til instruksjonene før bruk.2×PCR-blanding må pipetteres og blandes gjentatte ganger med en mikropipette før bruk for å unngå ujevn ionefordeling.
Legge merke til:
Ta ut manualen og les den nøye, og gjør forberedelser før eksperimentet i strengt samsvar med kravene i manualen.
Trinn 4
Forbered PCR-reaksjonssystem

I henhold til den eksperimentelle protokollen, bland primerne, H2O og 2×PCR-blanding jevnt, sentrifuger og fordel dem til hvert reaksjonsrør.
Legge merke til:
For storskala eller langtidstesting anbefales det å bruke et PCR-reaksjonssystem som inneholder UNG-enzym, som effektivt kan unngå aerosolforurensning forårsaket av PCR-produkter.
Trinn 5
Legg til reaksjonsmal

Ved å bruke Direct PCR-teknologi er det ikke behov for en kjedelig nukleinsyrerenseprosess, prøvemalen kan tilberedes innen 10 minutter, og det tilsvarende PCR-reaksjonssystemet kan tilsettes.
Legge merke til:
Spaltningsmetoden har bedre deteksjonseffekt, og det oppnådde produktet kan brukes til flere deteksjonsreaksjoner.

5.1: Direkte utvidelse av blader
I henhold til størrelsen på bildet i manualen, kutt bladvevet med en diameter på 2-3 mm og plasser det i PCR-reaksjonssystemet.
Merk: Sørg for at bladfragmentene er helt nedsenket i PCR-reaksjonsløsningen, og ikke tilsett overflødig bladvev.
5.2: Bladdelingsmetode
Skjær bladvevet med en diameter på 5-7 mm og plasser det i et sentrifugerør.Hvis du velger modne blader, vennligst unngå å bruke vevet i bladets hovedåre.Pipetter 50 ul buffer P1-lysat inn i et sentrifugerør for å sikre at lysatet kan nedsenke bladvevet fullstendig, plasser det i en termisk syklus eller et metallbad, og lysér ved 95°C i 5-10 minutter.

Tilsett 50 ul buffer P2 nøytraliseringsløsning og bland godt.Det resulterende lysatet kan brukes som en mal og legges til PCR-reaksjonssystemet.
Merk: Mengden av mal er mellom 5-10 % av PCR-systemet, og bør ikke overstige 20 % (for eksempel, i et 20μl PCR-system, tilsett 1-2μl lyseringsløsning, ikke mer enn 4μl).
Trinn 6
PCR-reaksjon

Etter sentrifugering av PCR-reaksjonsrøret, plasseres det i et PCR-instrument for amplifikasjon.
Legge merke til:
Reaksjonen bruker ikke-renset mal for amplifikasjon, så antallet amplifikasjonssykluser er 5-10 flere sykluser enn ved bruk av renset DNA-template.
Trinn 7
Elektroforesedeteksjon og resultatanalyse
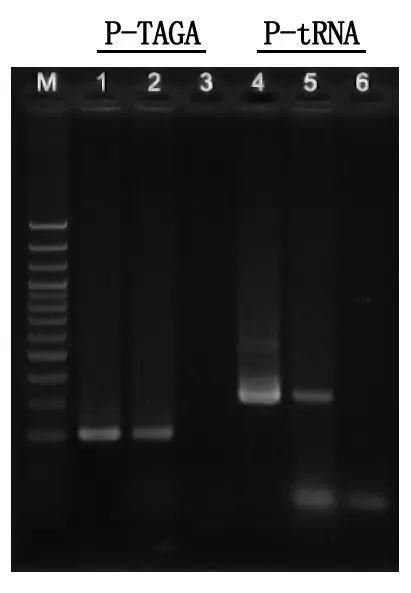
M: 100bp DNA-stige
1\4: Renset DNA-metode
2\5: Direkte PCR-metode
3\6: Tom kontroll
QC:
Testresultatene til de ulike kontrollene som er satt i eksperimentet, skal oppfylle følgende betingelser.Ellers bør årsaken til problemet analyseres, og testen bør utføres på nytt etter at problemet er eliminert.
Tabell 1. Normale testresultater av ulike kontrollgrupper
*Når plasmidet brukes som en positiv kontroll, kan resultatet av endogene gentest være negativt
Resultatvurdering:
A. Testresultatet for det endogene genet i prøven er negativt, noe som indikerer at DNA som er egnet for vanlig PCR-deteksjon ikke kan ekstraheres fra prøven eller at det ekstraherte DNAet inneholder PCR-reaksjonshemmere, og DNA bør ekstraheres på nytt.
B. Testresultatet for det endogene genet i prøven er positivt, og testresultatet for det eksogene genet er negativt, noe som indikerer at DNA egnet for vanlig PCR-deteksjon er ekstrahert fra prøven, og det kan bedømmes at XXX-genet ikke er påvist i prøven.
C. Testresultatet for det endogene genet i prøven er positivt, og testresultatet for det eksogene genet er positivt, noe som indikerer at DNA egnet for vanlig PCR-deteksjon er ekstrahert fra prøven, og prøve-DNAet inneholder XXX-genet.Bekreftelseseksperimenter kan utføres videre.
Trinn 8
Design deteksjonsprimere

Etter eksperimentet, bruk 2 % natriumhypoklorittløsning og 70 % etanolløsning for å tørke av eksperimentelle området for å forhindre miljøforurensning.
Innleggstid: sep-08-2021



